Résumé
L'isotherme d'adsorption chimique révèle des informations sur la surface active d'un matériau et est utilisé depuis de nombreuses années comme outil analytique standard pour l'évaluation des catalyseurs. Les techniques de réaction programmées en fonction de la température sont apparues dans les années 1950 comme un complément indispensable aux analyses de l'isotherme d'adsorption chimique dans de nombreux domaines de l'industrie et de la recherche. Cet article présente une introduction à ces techniques analytiques.
Introduction
La conception optimale et l'utilisation efficace des catalyseurs nécessitent une compréhension approfondie de la structure et de la chimie de surface du matériau catalytique. Les analyses d'adsorption chimique (chimisorption) peuvent fournir une grande partie des informations nécessaires à l'évaluation des matériaux catalytiques dans les phases de conception et de production, ainsi qu'après une période d'utilisation. L'équipement analytique requis peut être relativement peu coûteux, simple à utiliser et rapide par rapport à d'autres équipements capables d'obtenir les mêmes informations.
Différencier l'adsorption physique de l'adsorption chimique
Un matériau solide présente généralement une distribution hétérogène de l'énergie de surface. Les molécules de gaz, de vapeur ou de liquide peuvent se lier à la surface si elles s'en approchent suffisamment pour interagir. Les discussions de ce document se limitent à l'adsorption (et à la désorption) de gaz ou de vapeurs sur (ou à partir de) des surfaces solides. Le solide est appelé adsorbant; la molécule de gaz ou de vapeur avant d'être adsorbée est appelée adsorbant et, lorsqu'elle est liée à la surface solide, l'adsorbat.
L'adsorption physique est le résultat d'une interaction solide-gaz relativement faible. Il s'agit d'une attraction physique résultant de forces de Van der Waal non spécifiques et relativement faibles et d'une énergie d'adsorption ne dépassant généralement pas 80 kJ/mole, les énergies typiques étant considérablement inférieures. Les molécules adsorbées physiquement peuvent diffuser le long de la surface de l'adsorbant et ne sont généralement pas liées à un endroit spécifique de la surface. N'étant que faiblement liée, l'adsorption physique est facilement inversée.
L'adsorption peut également donner lieu à un complexe de surface, une union beaucoup plus forte qu'une liaison physique avec des chaleurs d'adsorption allant jusqu'à environ 600 kJ/mole pour les liaisons C-N et 800 kJ/mole pour les liaisons chimiques. Une liaison chimique implique le partage d'électrons entre l'adsorbat et l'adsorbant et peut être considérée comme la formation d'un composé de surface. En raison de la force de la liaison, l'adsorption chimique est difficile à inverser.
L'adsorption physique a lieu sur toutes les surfaces, à condition que les conditions de température et de pression soient favorables. La chimisorption, en revanche, est très sélective et ne se produit qu'entre certaines espèces adsorbantes et adsorbées et seulement si la surface chimiquement active est débarrassée des molécules précédemment adsorbées.
Dans des conditions appropriées, l'adsorption physique peut aboutir à la formation de plusieurs couches de molécules adsorbées. La chimisorption, dans le cas typique, ne se produit que tant que l'adsorbant peut entrer en contact direct avec la surface ; il s'agit donc d'un processus à une seule couche. Il peut y avoir des exceptions si l'adsorbant est très polaire, comme par exemple le NH3. L'adsorption physique et l'adsorption chimique peuvent se produire simultanément sur la surface ; une couche de molécules peut être physiquement adsorbée sur une couche chimiquement adsorbée sous-jacente. La même surface peut présenter une physisorption à une température donnée et une chimisorption à une température plus élevée. Par exemple, à la température de l'azote liquide (77 K), l'azote gazeux est adsorbé physiquement sur le fer, mais à 800 K, un niveau d'énergie trop élevé pour les liaisons d'adsorption physique, l'azote est adsorbé chimiquement pour former du nitrure de fer (Moore).
Applications générales de la sorption de gaz en tant qu'outil analytique
L'observation du processus d'adsorption permet de déterminer diverses caractéristiques de la surface. La quantité de molécules absorbées par une surface dépend de plusieurs variables, notamment la température, la pression, la distribution de l'énergie de surface, ainsi que la surface et la porosité du solide. La relation entre la quantité de molécules adsorbées et la pression à température constante est appelée isotherme d'adsorption.
Les isothermes d'adsorption et de désorption physiques sont importantes pour caractériser l'ensemble de la surface. Le moindre changement dans la forme de l'isotherme tracée est révélateur d'une caractéristique particulière de la surface. Les analyses des données des isothermes d'adsorption physique révèlent la surface totale, le volume et la surface des mésopores et des micropores, le volume total des pores, la distribution du volume et de la surface des pores en fonction de leur taille et la distribution de l'énergie de surface. L'adsorption physique est donc un outil important dans l'étude des catalyseurs, en particulier pour évaluer la structure du support.
L'isotherme d'adsorption chimique évalue également la surface, mais elle est sélective en ce sens qu'elle ne sonde que les zones actives, c'est-à-dire celles qui sont capables de former une liaison chimique avec le gaz ou la vapeur adsorbée. Cette sélectivité implique à la fois la molécule sonde et la composition moléculaire ou atomique de la matière active, de sorte que le choix des molécules sondes est beaucoup plus important dans les tests de chimisorption. Bien que les tests de chimisorption isotherme soient importants pour caractériser les surfaces actives, les tests programmés en fonction de la température le sont encore plus. La forme de base des données produites par ces tests ressemble à un chromatogramme de la température en fonction de la quantité désorbée.
Les bases du catalyseur
Un catalyseur affecte la vitesse d'une réaction chimique. Le mot "vitesse" est le plus important dans cette description, car un catalyseur ne peut pas induire une réaction qui n'est pas autorisée par les lois de la thermodynamique. Un catalyseur ne peut qu'augmenter la vitesse à laquelle la réaction s'approche de l'équilibre.
Les catalyseurs hétérogènes comprennent les métaux, les oxydes métalliques et les acides solides. Les métaux purs peuvent être utilisés comme catalyseurs solides ou être dispersés sous forme de petits grains à la surface d'un matériau de support tel que TiO2, ZrO2, Al2O3 ou SiO2. La préparation d'un catalyseur sur support implique la sélection de précurseurs des composants actifs et de tout promoteur nécessaire et leur mélange dans un solvant. Le mélange forme un précipitant, ou est utilisé pour recouvrir un support inerte, ou est utilisé pour imprégner un support. Finalement, le métal actif ou le précurseur est dispersé sur le support. Le produit est séché, mélangé à un liant ou à un agent de formation, puis broyé, granulé, extrudé ou mis en forme d'une autre manière. Enfin, le matériau est calciné et activé par oxydation, réduction ou autre.
Un type de catalyseur sans support est composé de métal pur. Les catalyseurs métalliques Raney, par exemple, sont préparés en dissolvant un alliage d'aluminium et de nickel dans une solution d'hydroxyde de sodium, qui dissout le composant aluminium. Le produit final est une "éponge" métallique active très poreuse, tous les autres matériaux d'échafaudage ayant été éliminés. Un autre catalyseur métallique sans support est produit par fusion ou frittage d'oxydes métalliques avec des promoteurs, formant ainsi un réseau de pores dans toute la masse métallique (van der Laan). Le catalyseur de synthèse de l'ammoniac à base de fer fondu en est un exemple.
Les catalyseurs zéolithiques forment un autre groupe. Les zéolithes sont des aluminosilicates hydratés largement utilisés dans l'industrie chimique et les opérations de raffinage. Leur activité est influencée par le rapport entre la silice et l'alumine. Les catalyseurs silice-alumine amorphes ont une activité plus faible que les catalyseurs zéolitiques et sont utilisés dans les opérations d'hydrocraquage doux. Une surface acide est nécessaire pour le craquage et l'acidité est associée aux atomes d'oxygène attachés aux atomes d'aluminium.
Chimisorption et catalyseurs
Le processus catalytique peut être généralisé par une courte séquence d'étapes comme suit. Considérons les molécules réactives A et B dans la masse gazeuse au-dessus d'une surface solide qui supporte un ensemble de sites métalliques actifs S. Si la molécule A est chimiquement adsorbée sur l'un des sites actifs, un complexe de surface A est formé. Ensuite, A réagit avec B en formant la molécule A+B, qui s'échappe du site, régénérant ainsi le site S.
La sorption d'au moins une des molécules réactives est nécessaire pour que la catalyse se produise. Si l'accélération de la réaction était simplement due à la concentration de molécules à la surface, la catalyse résulterait de l'adsorption physique des réactifs. La chimisorption est une étape essentielle, la molécule adsorbée formant un complexe de surface intermédiaire plus réceptif à la réaction chimique. La dépendance de la catalyse à l'égard de la chimisorption est l'une des raisons pour lesquelles la chimisorption est une technique analytique si instructive dans l'étude de la catalyse : la chimie qui se produit lors de l'application du catalyseur est observée directement en laboratoire.
Les performances d'un catalyseur dépendent de plusieurs variables. Tout d'abord, les sites d'adsorption doivent être à la fois nombreux et disponibles pour les molécules réactives. Dans certains cas, les grains de métal actif se trouvent à la surface, mais il y a aussi des grains situés sous la surface et inaccessibles aux réactifs. Le simple fait que les sites d'adsorption soient situés à la surface ne suffit pas à garantir des performances optimales. Par exemple, certains sites d'adsorption potentiels peuvent être situés profondément dans un micropore trop étroit pour que la molécule de réactif puisse y pénétrer ou que le produit de la réaction puisse en sortir ; dans ce cas, le site de surface ne peut pas participer activement à la chimisorption. Un site peut être situé le long d'un chemin tortueux qui empêche l'écoulement efficace des réactifs vers le site actif et des produits loin du site. Étant donné que de nombreux métaux actifs sont très coûteux, un critère de conception important consiste à maximiser le nombre de sites actifs par unité de métal.
L'activité catalytique dépend de la rapidité de la chimisorption et de la force (énergie) de la liaison de chimisorption. Si la liaison est trop faible, la molécule peut se désorber avant de réagir ; si elle est trop forte, la libération du produit et la régénération du site peuvent être retardées. Les méthodes de chimisorption isotherme et les méthodes de chimisorption programmée en fonction de la température peuvent être utilisées pour étudier la distribution de l'énergie de surface.
La chimisorption décrite ci-dessus concerne l'utilisation de catalyseurs dans diverses applications. Les mêmes réactions, mais à plus petite échelle, peuvent également se produire dans un tube à échantillon dans des conditions contrôlées, ce qui permet d'étudier le processus. Il s'agit de la chimisorption utilisée comme technique analytique. Les analyses de chimisorption sont appliquées pour caractériser physiquement un matériau catalytique, pour déterminer l'efficacité relative d'un catalyseur dans la promotion d'une réaction particulière, pour étudier l'empoisonnement du catalyseur et pour surveiller la dégradation de l'activité catalytique au fil du temps. Parmi les diverses techniques instrumentales utilisées pour analyser les catalyseurs, les techniques de chimisorption sont les plus universellement employées.
Instruments de chimisorption
Les analyses de chimisorption isotherme sont obtenues par deux techniques de chimisorption : a) la chimisorption volumétrique statique et b) la chimisorption dynamique (gaz en circulation).
La technique volumétrique permet d'obtenir une mesure à haute résolution de l'isotherme de chimisorption à partir d'une pression très basse jusqu'à la pression atmosphérique, à n'importe quelle température, depuis la température ambiante jusqu'à plus de 1000 oC. Les applications commerciales de cette technique sont presque exclusivement automatisées. L'obtention d'une isotherme à haute résolution nécessite de nombreuses étapes de dosage précises pour atteindre le point d'équilibre, ainsi que de nombreuses étapes de pression qui, sans automatisation, constitueraient une procédure longue et sujette aux erreurs.
La technique du gaz en circulation (dynamique) fonctionne à la pression ambiante. Après nettoyage de l'échantillon, de petites injections (doses) de quantités connues avec précision d'adsorbant sont administrées par impulsions jusqu'à ce que l'échantillon soit saturé, d'où le nom de "chimisorption par impulsions". Un détecteur de conductivité thermique étalonné (TCD) contrôle la quantité d'adsorbant qui n'est pas absorbée par les métaux actifs. Cette quantité est soustraite de la quantité injectée, ce qui donne la quantité adsorbée à chaque injection. Ces quantités sont additionnées pour déterminer la capacité de la masse de l'échantillon. Les injections peuvent être effectuées à l'aide d'une seringue ou d'une vanne d'injection en boucle, manuelle ou automatisée, de type HPLC. Étant donné que le point final (saturation) est le seul point mesuré, le nombre d'injections peut être faible et il n'y a pas de changement de pression. C'est pourquoi la technique des gaz en écoulement est souvent réalisée manuellement, bien qu'il existe également des instruments automatisés.
Au cours des dernières décennies, les analyseurs de chimisorption programmée en température ont été reconnus comme des outils extrêmement précieux pour la caractérisation des catalyseurs. La méthode est parfois simplement appelée réactions programmées en fonction de la température et comprend la désorption programmée en fonction de la température (TPD), la réduction programmée en fonction de la température (TPR) et l'oxydation programmée en fonction de la température (TPO). La méthode de chimisorption dynamique est particulièrement adaptée à la réalisation d'analyses de programmes de température.
Analyses de chimisorption
La polyvalence de la technique de chimisorption est illustrée par l'étendue des informations qu'il est possible d'obtenir sur un matériau. Une partie de cette polyvalence a été révélée dans la discussion ci-dessus. Dans les sections suivantes, d'autres possibilités sont explorées.
Dans les exemples qui suivent, seuls les catalyseurs composés d'une seule espèce active sont pris en compte pour simplifier les calculs. Dans le cas de catalyseurs métalliques mixtes, de nombreuses équations ci-dessous nécessiteraient d'additionner une série de termes, un pour chaque espèce adsorbante et chaque terme pondéré par la contribution fractionnelle de l'espèce à l'ensemble.
L'isotherme de chimisorption
Une isotherme est un ensemble de points de données relatifs à la quantité adsorbée en fonction de la pression qui caractérisent le processus d'adsorption à une température constante. Pour mieux comprendre, considérons un réacteur de volume constant V qui contient un échantillon et une quantité de molécules de gaz N. La théorie de la chimisorption suppose que la surface active d'un solide contient un nombre fixe Ns de sites d'adsorption et qu'une seule molécule à la fois peut occuper un site. Les molécules présentes dans le tube à échantillon qui sont en compétition pour un site d'adsorption représentent une concentration C qui peut être exprimée en général par la résolution de la loi des gaz, soit
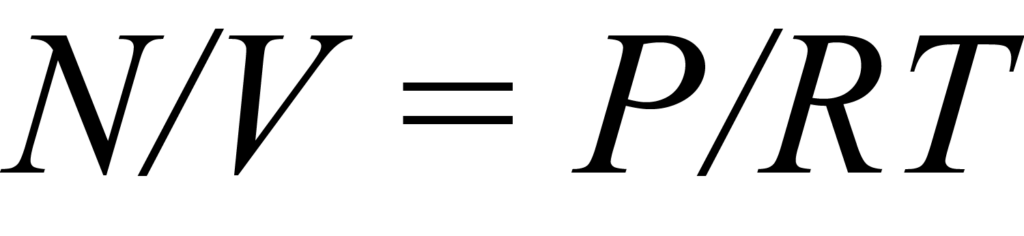
où N et V ont été définis précédemment, P est la pression, T la température et R la constante des gaz.
Pour un système gaz-solide spécifique à l'équilibre dans des conditions spécifiques de température et de pression, une certaine fraction ϑ du nombre total de sites disponibles Ns sera occupée par nads molécules adsorbées. Ce phénomène est décrit par
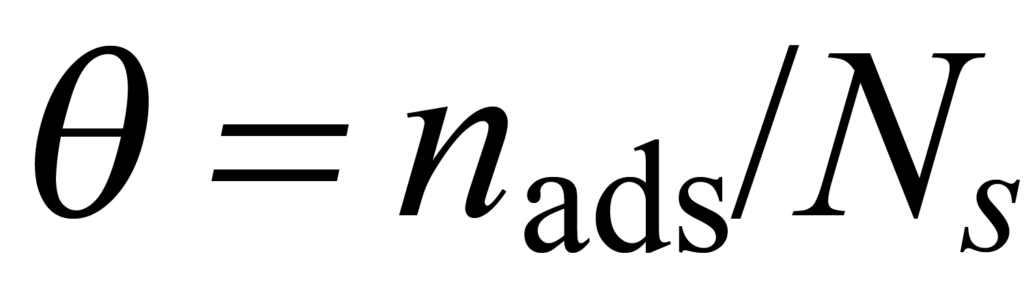
La vitesse à laquelle l'équilibre est atteint dépend de la concentration des molécules impliquées ainsi que d'autres considérations, ces dernières étant regroupées dans un paramètre appelé "constante de vitesse" k.
Pour le processus d'adsorption, la concentration des molécules non adsorbées en compétition pour les sites restants est C-ϑC et le taux d'adsorption est kads(C-ϑC) ou kadsC(1-ϑ). Pour la désorption, la fraction de molécules désorbées est ? et la vitesse de désorption est kdesϑ. Lorsque l'équilibre est atteint, les taux d'adsorption et de désorption sont égaux, c'est-à-dire,
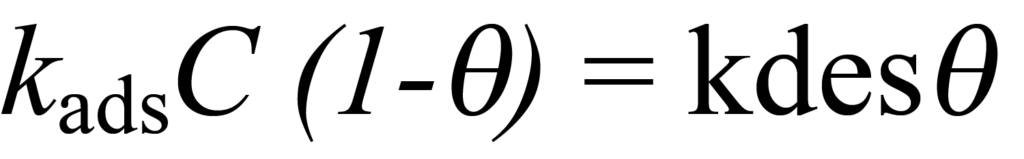
ou
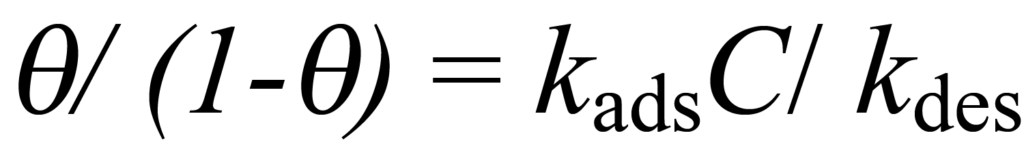
En utilisant K pour représenter les kads/kdes et en résolvant pour ϑ, on obtient
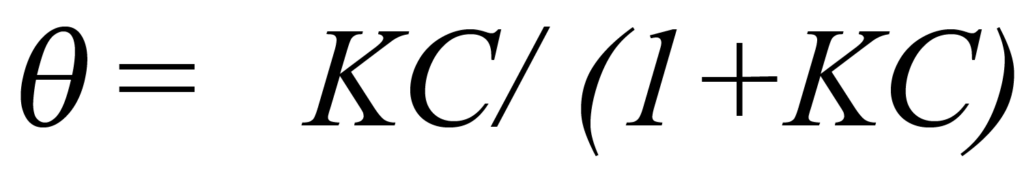
Étant donné que l'environnement est de volume constant V et de température constante T, la modification de la pression est le seul moyen de modifier la concentration C. L'équation 1 établit que la concentration est directement proportionnelle à la pression. Par conséquent, si la constante de proportionnalité concentration-pression est combinée à K et représentée par b, l'équation 4 peut s'écrire
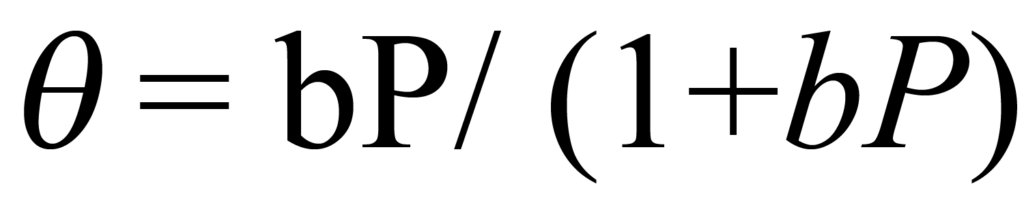
Il s'agit de la célèbre isotherme de Langmuir, qui décrit avec précision la plupart des isothermes de chimisorption n'impliquant pas de fragmentation (dissociation) de la molécule adsorbante. L'isotherme est asymptotique à ϑ = 1, ce qui correspond à une couverture totale ou à l'achèvement de la monocouche.
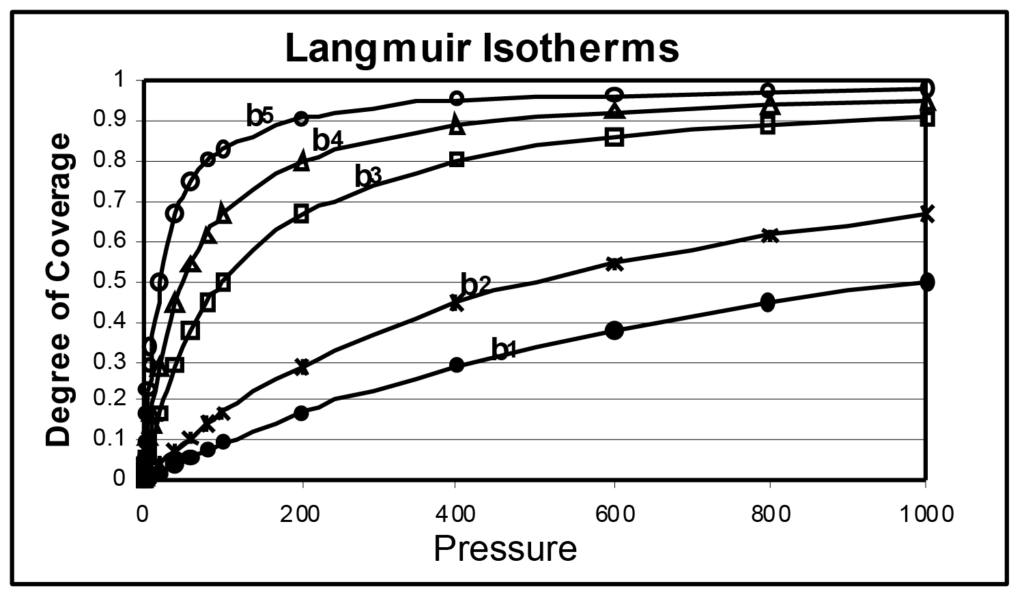
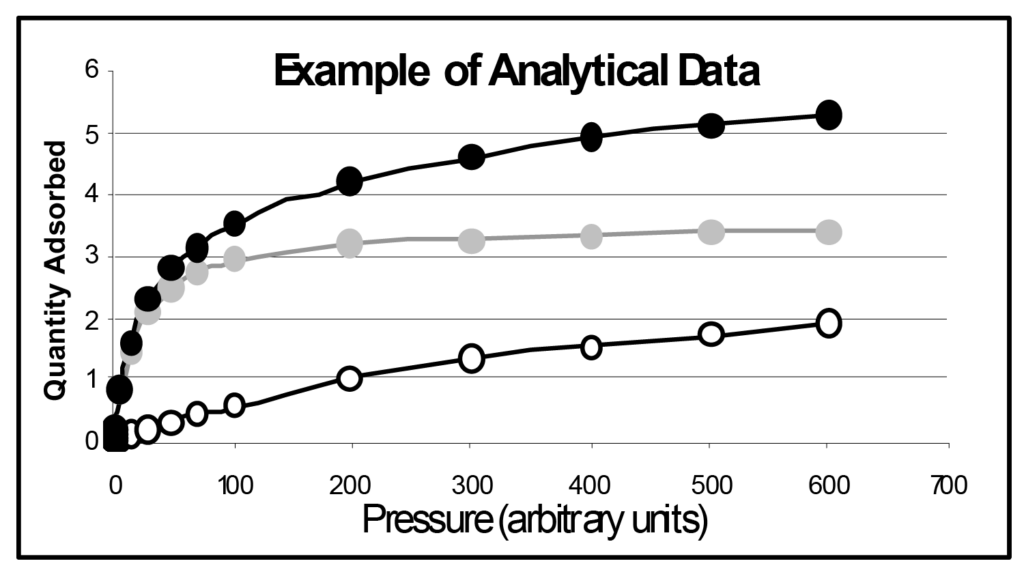
Plusieurs isothermes de Langmuir sont illustrées dans la figure 1, la seule différence étant la magnitude de b. Le paramètre b est directement lié à l'énergie de surface ; l'augmentation de l'énergie de surface augmente la probabilité d'adsorption à une pression donnée. Le paramètre b est inversement lié à la température qui, lorsqu'elle augmente, accroît l'énergie moléculaire et diminue la probabilité d'adsorption à une pression donnée.
Dans l'illustration, b1 est inférieur à b2, qui est inférieur à b3, et ainsi de suite, b5 étant 50 fois supérieur à b1. On peut en déduire que le même échantillon analysé à cinq températures différentes produirait un ensemble similaire d'isothermes, tout comme cinq échantillons différents ayant une énergie de surface différente et mesurés à la même température d'analyse.
L'obtention d'une isotherme de chimisorption n'est pas nécessairement une mesure directe, comme l'illustrent les figures 2. Dans le cas typique indiqué par ces illustrations, il y aura des molécules (indiquées par R) faiblement adsorbées à la surface du support et il y aura également une monocouche de molécules chimisorbées (indiquées par I) sur la surface active. En fonction de la pression et de la température, des molécules peuvent également être physiquement adsorbées sur la monocouche chimisorbée.
L'adsorption faible est appelée adsorption réversible et la chimisorption forte, irréversible, d'où les étiquettes R et I dans l'illustration. L'isotherme initial sera une combinaison d'adsorption réversible et irréversible.
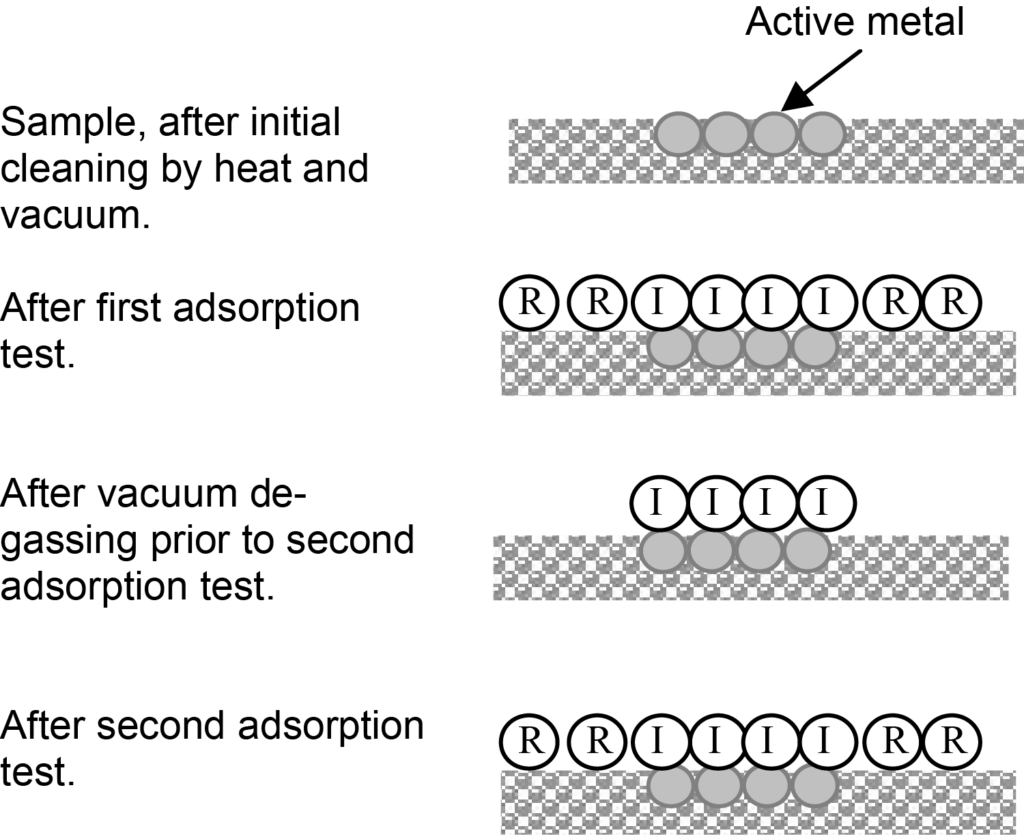
Les contributions réversibles et irréversibles à une isotherme combinée peuvent être distinguées en effectuant un second test d'adsorption. Après le test d'adsorption initial et avant le second test, l'échantillon est soumis au vide uniquement, ce qui entraîne la désorption des molécules faiblement adsorbées et ne laisse que les molécules qui ont formé une liaison de chimisorption forte avec la surface active.
Le deuxième test d'adsorption est réalisé dans les mêmes conditions que le test initial, mais cette fois la surface active est déjà recouverte d'une monocouche chimisorbée. L'absorption de l'adsorbat sera uniquement celle associée à l'adsorption réversible. En soustrayant la quantité adsorbée de manière réversible de l'isotherme combinée à chaque valeur de pression, on obtient une isotherme d'adsorption irréversible. Les résultats analytiques sont illustrés dans la figure 3.
Nombre de sites actifs accessibles
Les techniques de chimisorption statique volumétrique et dynamique peuvent être utilisées pour mesurer la quantité de gaz nécessaire à la formation d'une monocouche de chimisorbat sur une surface active. La méthode dynamique ne fournit qu'un seul point de données, qui correspond à la quantité d'adsorbant nécessaire pour saturer la surface active avec une monocouche. La capacité monocouche de la surface active est calculée à partir de l'isotherme d'adsorption irréversible dérivée illustrée dans la figure 3. Étant donné que l'isotherme d'adsorption irréversible est asymptotique à la valeur de la couverture totale (de la surface active), le plateau de l'isotherme est extrapolé sur l'axe des y pour obtenir la valeur de la capacité monocouche. L'extrapolation sur l'axe des ordonnées de la section linéaire des isothermes réversibles-irréversibles combinées fournit également une valeur raisonnable pour la quantité de couverture monocouche.
La méthode de chimisorption dynamique consiste à injecter (manuellement ou automatiquement) de petites quantités d'adsorbant, connues avec précision, dans un flux de gaz porteur inerte qui traverse le lit de l'échantillon.
La pression partielle de l'adsorbant augmente momentanément sur l'échantillon lors du passage de l'impulsion et une partie ou la totalité de la quantité de gaz injectée est adsorbée. Le gaz non adsorbé est balayé par le détecteur où il est enregistré. Il convient de noter que, contrairement à la méthode statique (décrite ci-dessus), l'adsorption nette n'est qu'une adsorption irréversible car, après le passage de l'impulsion d'adsorbant, la pression partielle de l'adsorbant dans le gaz en vrac est essentiellement nulle et les molécules faiblement adsorbées qui s'échappent sont entraînées vers le détecteur par le flux porteur.
Au fur et à mesure que la monocouche se forme sur la surface active, l'échantillon absorbe de moins en moins l'adsorbant injecté. Les injections se poursuivent jusqu'à ce que l'échantillon soit saturé, comme l'indique le fait que tous les pics suivants du détecteur restent de la même taille. Les résultats d'un tel test sont présentés dans la figure 4, qui illustre la sortie du détecteur. La surface de chaque pic est proportionnelle à la quantité de l'injection qui n'a pas été adsorbée.
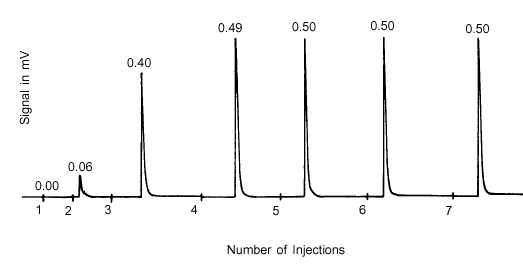
Dans la figure 4, la surface intégrée des pics associés aux injections 5, 6 et 7 indique qu'il n'y a pas eu d'adsorption et que la zone active est donc saturée. La totalité de la première injection a été adsorbée, de même que 88 % de l'injection 2, 20 % de l'injection 4 et 2 % de l'injection 3. À partir de la quantité connue de chaque injection, de la fraction de chaque injection qui a été adsorbée et de la masse de l'échantillon, on détermine le nombre de moles de gaz adsorbant absorbées par chaque gramme d'échantillon.
Le processus d'adsorption peut impliquer la séparation d'un adsorbant moléculaire en deux ou plusieurs entités moléculaires ou atomiques (ions ou radicaux), chaque entité formant une liaison avec des sites de surface actifs individuels. On parle alors d'adsorption dissociative ou de second ordre, par opposition à l'adsorption de premier ordre ou non dissociative, dans laquelle la molécule adsorbante reste intacte pendant l'adsorption. Le résultat de l'adsorption dissociative est que le nombre de molécules adsorptivesNm déterminées comme ayant été absorbées par gramme d'échantillon n'est pas égal au nombre d'atomes de la surface active qui ont participé à la chimisorption.
Pour déterminer le nombre d'atomes de surface impliqués, il faut tenir compte de la stœchiométrie de la réaction de surface. La stœchiométrie fait référence à la relation entre les quantités de substances qui réagissent ensemble dans une réaction chimique particulière et les quantités de produits qui sont formés. Par exemple, une molécule d'hydrogène (H2) peut se dissocier en deux atomes d'hydrogène et réagir avec deux atomes de la surface active, comme c'est le cas avec le Pt. Ainsi, le nombre de molécules adsorbantes absorbées par la surface active doit être multiplié par un facteur de stœchiométrie Fs (où Fs = 2 dans le cas de l'exemple) pour obtenir le nombre d'atomes de la surface Ns ou "sites". Mathématiquement,
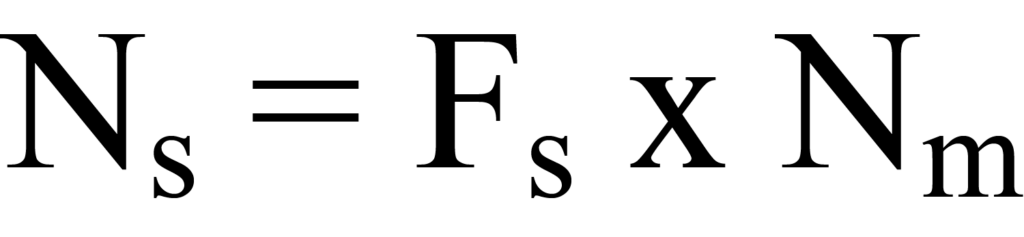
où Ns etNm sont déterminés par gramme d'échantillon.
La molécule adsorbante peut être capable de se lier dans plus d'une configuration avec la surface active. La proportion de molécules se liant de chaque manière possible doit être prise en considération, ce qui donne souvent un facteur de stœchiométrie non entier.
Surface active
La surface active spécifique résulte du nombre de molécules Ns adsorbées sur la surface active d'un gramme d'échantillon. La surface active spécifiqueAA est déterminée en multipliant la surfaceAm occupée par une molécule de surface par le nombre de molécules adsorbées par gramme Ns. La surface occupée par un seul atome provenant de l'adsorption peut généralement être trouvée dans la littérature. Elle peut également être déterminée expérimentalement, si nécessaire, en déterminant la surface BET d'un échantillon de matière active pure à l'aide de N2, puis en déterminant l'absorption molaire de l'adsorbant actif sur le même échantillon.
Dispersion des métaux et pourcentage de métal
La dispersion du métal décrit le rapport entre le nombre d'atomes de métal actif disponibles pour la réaction et le nombre total d'atomes de métal dans le matériau catalytique. La quantité de métal actif incorporé par unité de masse de matériau de support est disponible à partir de la formule de fabrication ; cela donnera la quantité d'atomes de métal actifNT par unité de masse de catalyseur. L'analyse de chimisorption décrite ci-dessus est utilisée pour déterminer la quantité de métal actif par gramme disponible pour la réaction. Le pourcentage de dispersion est le rapport entre la quantité disponible et la quantité totale de molécules actives multiplié par 100 %, soit
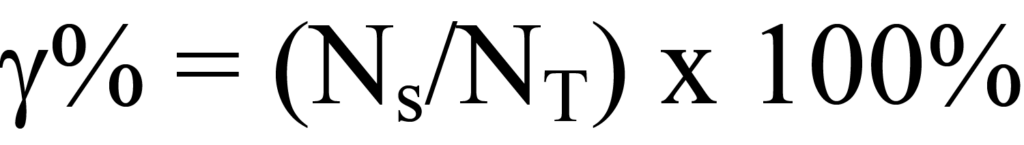
La dispersion est exprimée comme un rapport entre le métal disponible et le métal total. Une autre fraction déterminée à partir de la procédure de fabrication est le rapport entre le poids du métal et la masse du matériau catalytique en vrac, exprimé sous la forme d'une fraction décimale ou d'un pourcentage.
Taille des particules actives (cristallites)
Cette estimation de la taille des particules actives est un calcul géométrique basé sur l'hypothèse que la forme des cristallites est de géométrie régulière, une sphère étant généralement la géométrie choisie. Les calculs précédents ont permis de connaître la surface active par gramme d'échantillonAA et, grâce à la procédure de fabrication du catalyseur, la proportion fractionnelle de métal dans la masse du catalyseur.
Le calcul utilise une expression de la géométrie du grain. À partir de la géométrie régulière supposée, le diamètre peut être exprimé en termes de surface et de volume. Le volume de métal actif n'est pas connu, alors que la densité atomique ou moléculaire ρm l'est ; le volume peut donc être exprimé en termes de densité. La surface de métal actif par gramme d'échantillon a été mesurée précédemment, ce qui a permis d'obtenir une valeur pour la surface par unité de masse de métalAm (m2/g). En substituant ces expressions à la relation générale D = f(A,V), on obtient
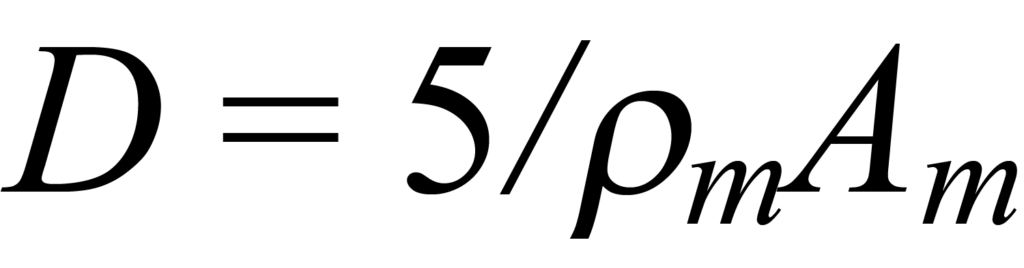
pour un grain cubique en surface (cinq des six faces exposées) et
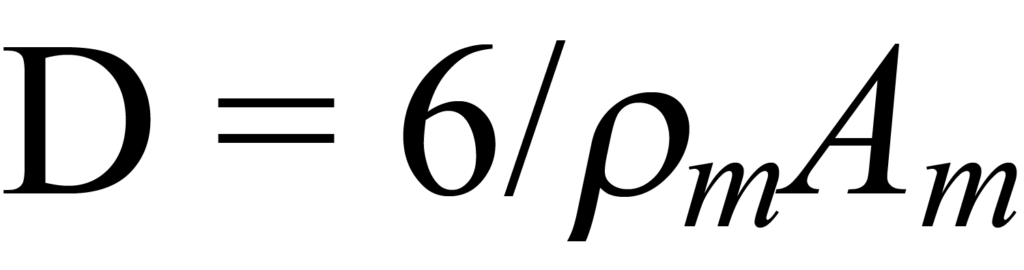
pour une particule de forme hémisphérique.
Le diamètre calculé par l'équation ci-dessus représente le diamètre moyen des grains de métal actif sur lesquels l'adsorption s'est produite.
Chimisorption programmée en fonction de la température
Comme son nom l'indique, la chimisorption programmée en fonction de la température est une méthode qui implique les effets de la température sur les réactions de surface. Trois réactions principales sont étudiées en fonction de la température : 1) la désorption, 2) la réduction et 3) l'oxydation.
La désorption programmée en fonction de la température par la méthode dynamique consiste à placer un échantillon dans une cellule d'échantillonnage et à le prétraiter pour éliminer toutes les espèces adsorbées de la surface active. Ensuite, un gaz ou une vapeur sélectionnés sont chimisorbés sur les sites actifs jusqu'à saturation, après quoi les molécules adsorbées restantes sont éliminées à l'aide d'un gaz inerte. La température (énergie) est augmentée à un rythme contrôlé tandis qu'un flux constant de gaz inerte est maintenu au-dessus de l'échantillon. Le gaz inerte et les molécules désorbées sont contrôlés par un TCD. Le signal TCD est proportionnel à la quantité de molécules désorbées lorsque l'énergie thermique, contrôlée par un thermocouple, surmonte l'énergie de liaison. Les quantités désorbées à des températures spécifiques fournissent des informations sur le nombre, la force et l'hétérogénéité des sites d'adsorption. Les données d'analyse sont généralement représentées sous forme de quantité désorbée en fonction de la température, ou sous forme de température et de quantité désorbée en fonction du temps.
La figure 5 est un profil TPD typique de la désorption de l'hydrogène à partir de Pt supporté sur Al2O3. Le premier pic est obtenu à une température de 393 K. Ce pic correspond à la faible adsorption d'hydrogène et peut être lié à la désorption du support ou à une faible chimisorption. Le deuxième pic obtenu à la température immédiatement supérieure (493 K) correspond probablement à une fuite d'hydrogène due à la présence de Pt sur l'alumine. Le troisième et dernier pic obtenu à 570 K correspond à l'hydrogène chimisorbé par le Pt. Ce pic est quantifié pour estimer la quantité de Pt disponible pour l'activité catalytique dans un réacteur. Les énergies de liaison peuvent être quantifiées à l'aide de la méthode TPD, comme nous le verrons plus loin.

La réduction programmée en fonction de la température est une méthode par laquelle un mélange de gaz réducteur, tel que l'hydrogène dilué dans un gaz inerte, s'écoule sur un échantillon d'oxyde. La température initiale est généralement inférieure à la température de réduction. Ensuite, la température de l'échantillon est augmentée à un rythme constant et, lorsque la réduction commence, l'hydrogène est consommé à partir du mélange porteur, ce qui est détecté par un TCD. Lorsque la réduction cesse, il n'y a plus de consommation d'hydrogène et la conductivité thermique du gaz provenant du tube d'échantillonnage revient à la ligne de base. Plusieurs pics de réduction peuvent être détectés au cours de la rampe de température, car la réduction sera probablement amorcée à différents niveaux d'énergie thermique. Chaque pic correspond alors à un oxyde différent et l'amplitude de chaque pic est proportionnelle à la vitesse de réaction.
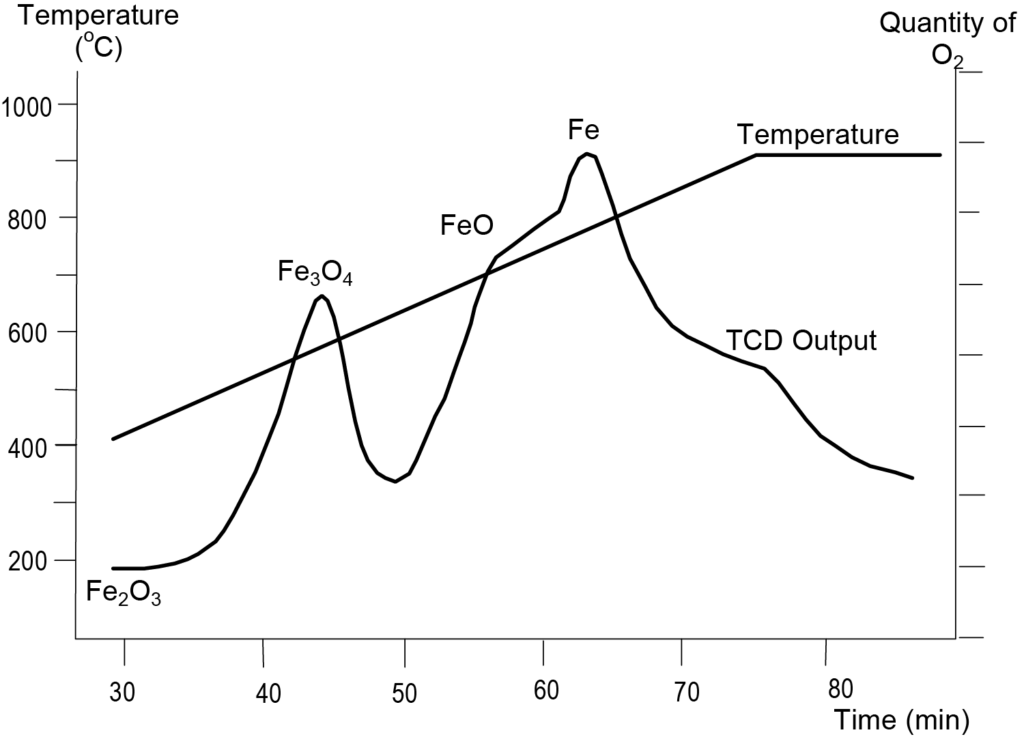
L'oxyde de fer sous forme d'hématite présente trois phases de réduction correspondant aux trois oxydes de fer. Une analyse TPR de l'hématite a été réalisée en utilisant un gaz réducteur composé de 10% d'hydrogène dans de l'argon et une vitesse de rampe de température de 10 °C/min. La figure 6 montre les résultats. Le premier pic de réduction apparaît à 575 K, correspondant à la transition de Fe2O3 à Fe3O4. Le pic à 627 K correspond à la transformation de Fe3O4 en FeO. Le dernier pic à 748 K correspond à la transition de FeO en Fe. Les positions des maxima montrés dans la figure 6 peuvent différer quelque peu d'un échantillon à l'autre en fonction de la taille des particules de Fe2O3et d'autres paramètres tels que le taux de rampe de température.
L'oxydation programmée en température peut être utilisée pour déterminer la quantité d'espèces réduites (également appelée degré de réduction), mais la difficulté de ce type d'analyse est d'atteindre l'oxydation totale. Plus couramment, la TPO est utilisée dans des applications telles que l'étude de la cinétique de la cokéfaction, l'évaluation de la combustion du carbone du catalyseur, la détermination des différentes formes de dépôts carbonés présents sur les catalyseurs après une réaction de décomposition du CO ou, plus généralement, les mesures de la consommation d'oxygène et des rendements en produits.
L'échantillon est chauffé à une vitesse uniforme tandis que le gaz réactif, généralement de 2 à 5 % d'oxygène, est appliqué à l'échantillon par impulsions, ou alternativement, sous forme de flux régulier. La réaction d'oxydation se produit à une température spécifique et entraîne une absorption d'oxygène. La quantité d'oxygène consommée au cours de la réaction est liée à la quantité d'une espèce sur la surface.
Énergie de surface et cinétique du premier ordre
Les mécanismes d'adsorption et de désorption sont composés d'une combinaison d'étapes cinétiques élémentaires. Par exemple, une molécule diatomique peut être adsorbée ou, lorsqu'elle s'approche de la surface, se fragmenter en atomes qui sont adsorbés indépendamment. Le premier cas est appelé chimisorption non dissociative ou cinétique de premier ordre, le second est la chimisorption dissociative ou cinétique de second ordre. La cinétique de premier ordre sera au centre des discussions suivantes.
L'interaction d'une molécule isolée avec une surface peut être représentée par un graphique de l'énergie potentielle en fonction de la distance par rapport à la surface, comme le montre la figure 7. À titre de comparaison, la figure 7 comprend également un tracé de l'énergie pour la physisorption ainsi que le chemin énergétique net emprunté par la molécule chimisorbée. En examinant ce diagramme, il faut comprendre qu'il s'agit d'une représentation de base d'une molécule unique s'approchant d'une surface et qu'il ne prend en compte que l'énergie en fonction de la distance par rapport à la surface, en négligeant les autres variables.
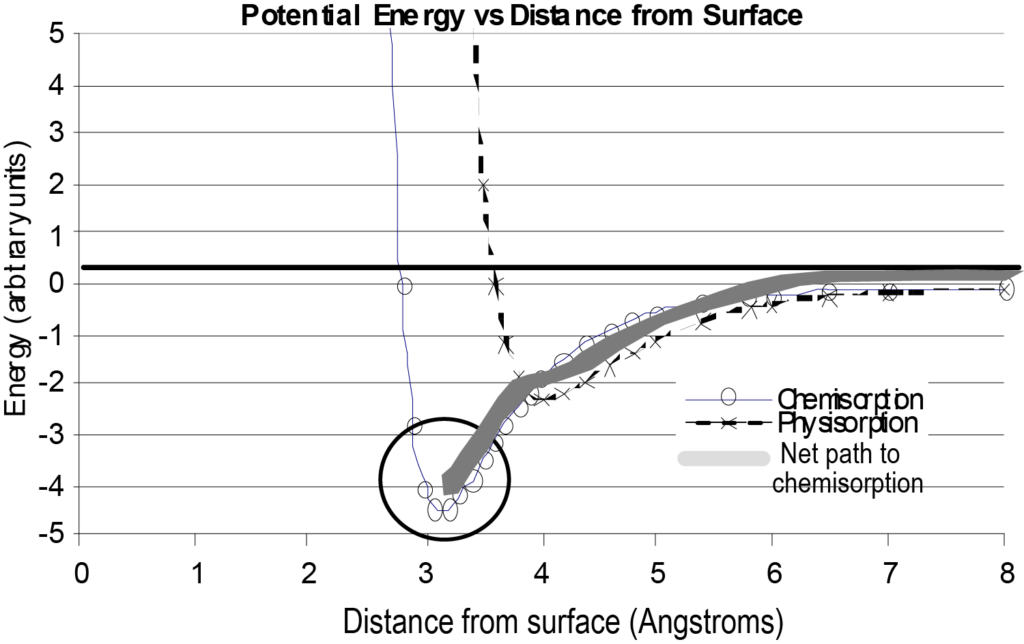
Une situation qui modifierait considérablement la forme de la courbe de chimisorption de la figure 7 serait que le processus d'adsorption soit dissociatif. La chimisorption dissociative nécessite une dépense d'énergie (énergie de dissociation) pour briser la molécule adsorbante. Le chemin de l'énergie nette présenterait alors un pic positif juste avant le puits d'énergie potentielle négatif. Ceci est illustré dans la figure 8. Comme dans la figure 8, le chemin d'adsorption physique est initialement plus favorable (énergie plus faible) lorsque la molécule s'approche de la surface, mais pour se dissocier et se chimisorber, une étape d'énergie positive est nécessaire pour effectuer la transition vers le chemin de chimisorption. Cette énergie est supérieure à l'énergie potentielle dissipée de la molécule.
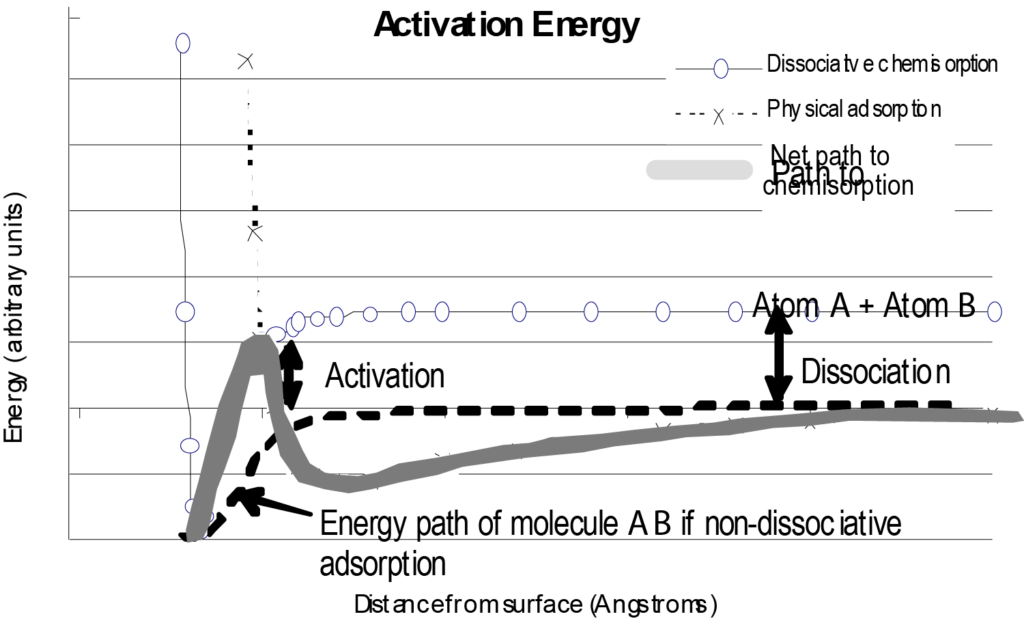
Une variable importante qui modifie la forme de la courbe d'énergie de chimisorption non dissociative est le nombre de molécules déjà adsorbées sur la surface. Les sites les plus énergétiques (les puits de potentiel les plus profonds) sont occupés en premier. Ce mode de couverture se poursuit jusqu'à ce que, finalement, les sites ayant l'énergie la plus faible soient occupés. L'énergie représentée en fonction de la couverture de la surface (charge) caractérise le degré d'hétérogénéité de l'énergie de surface. Ce graphique fournit des informations précieuses sur l'activité du catalyseur dans des conditions spécifiques. Pour certaines applications, plutôt que de tracer la distribution de l'énergie de surface, une seule valeur de l'énergie associée à la réaction peut suffire.
L'adsorption est un processus exothermique, qui finit donc par produire de l'énergie(énergie d'adsorption), même si un certain apport d'énergie(énergie d'activation) est nécessaire pour initier le processus. L'inversion de l'adsorption nécessite un apport d'énergie, la quantité requise étant la profondeur du puits de potentiel.
Les figures 7 et 8 concernent l'énergie potentielle d'une seule molécule isolée, mais c'est le potentiel énergétique de toutes les particules du système qui détermine si le système est en équilibre. À l'équilibre, une molécule située à n'importe quel endroit du système doit avoir le même potentiel énergétique ; cela s'applique aux molécules du gaz ou de la vapeur en vrac entourant le solide et aux molécules adsorbées à la surface du solide.
Pour un système adsorbat-adsorbant spécifique, l'équilibre d'adsorption est atteint par un équilibre délicat entre la pression P de la phase gazeuse, la température analytique T et le degré de couverture de la surface ϑ. Expérimentalement, l'un de ces paramètres est maintenu constant, une valeur est attribuée à l'autre et le troisième est observé pour déterminer sa valeur à l'équilibre. Une isotherme exprime le degré de couverture de la surface en fonction de la pression à température constante (ϑ = f(P)T), une isobare d'adsorption exprime le degré de couverture de la surface en fonction de la température à pression constante (ϑ = f(T)P), et une isostère montre la relation entre la pression et la température pour un degré de couverture constant. (P = f(T)ϑ).
L'équation de Clausius-Clapeyron exprime la chaleur d'adsorption à un degré spécifique de couverture de surface ϑ en termes de pression et de température, ce qui donne la chaleur d'adsorption isostérique qst. L'équation sous forme de différentielle partielle est la suivante
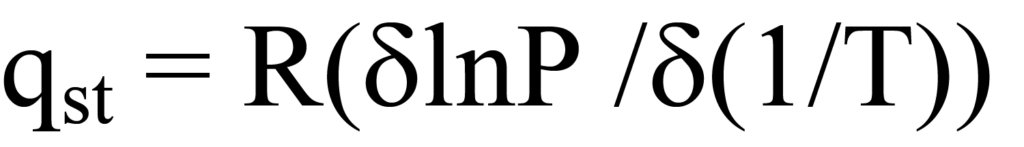
où T est la température, P la pression et R la constante des gaz.
Pour obtenir des valeurs connexes de température et de pression pour un degré de couverture donné, il faut obtenir plusieurs isothermes du même échantillon à différentes températures et mettre à l'échelle l'axe de la quantité adsorbée en unités de degrés de couverture. Cela permet d'extraire de l'ensemble des isothermes un ensemble d'isobares représentant le degré de couverture en fonction de la température sur la plage de température utilisée dans les analyses. À partir de l'ensemble des isobares, il est possible d'extraire un ensemble de valeurs de pression en fonction de la température pour diverses valeurs de volume adsorbé. Ces valeurs sont représentées sous forme de ln(P) en fonction de 1/T.
L'équation de Clausius-Clapeyron, ci-dessus, peut être regroupée sous forme linéaire, y = m(x), ce qui donne
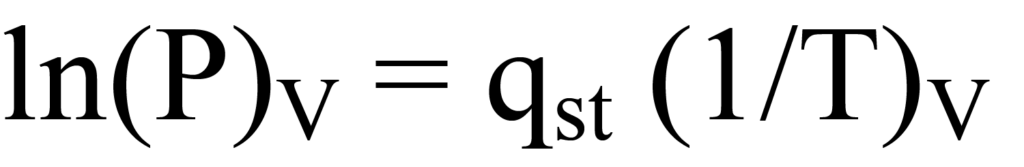
R étant une constante d'échelle qui n'affecte pas la pente. Pour chaque valeur de la quantité adsorbée, il existe une pente différente, qst. En traçant l'ensemble des pentes en fonction de la quantité adsorbée, on obtient le graphique recherché de l'énergie de surface en fonction de la quantité adsorbée, qui se rapporte à la couverture de la surface ou à la charge.
La chaleur d'adsorption peut également être calculée à partir des données obtenues par chimisorption dynamique par ce qui est parfois appelé la méthode de "variation du taux de chauffage". Étant donné que la quantité adsorbée en fonction de la pression n'est pas contrôlée par la méthode dynamique, il faut chercher une expression de l'énergie autre que celles des équations 10 et 11. A titre d'exercice, une telle expression est dérivée dans les paragraphes suivants. Le résultat (équation 21) peut être pris et utilisé sur la foi et l'on peut s'épargner le travail fastidieux des maths. Cependant, la dérivation étape par étape est présentée ici parce qu'elle peut intéresser certains lecteurs et qu'une dérivation détaillée est difficile à trouver dans la littérature. Des parties de ce qui suit ont été glanées dans les travaux de Schroeder et Gottfried, Houston, Nix et Garrett.
La désorption implique le taux de variation temporelle du nombre de molécules adsorbées N, exprimé algébriquement par ΔN/Δt. Cela peut également être considéré comme la vitesse de variation temporelle du nombre de sites d'adsorption disponibles Na.
(Note : Les changements dépendant du temps et de la température sont plus facilement traités mathématiquement en considérant que chaque pas de changement de temps ou de température est infiniment petit. Ainsi, DN/Dt peut être remplacé par ΔN/Δt et la manipulation algébrique est remplacée par le calcul différentiel. Pour les lecteurs non familiarisés avec le calcul, toute expression de la forme dX/dY peut être considérée comme la pente d'un tracé de X en fonction de Y à une valeur donnée de X).
L'expression cinétique de la vitesse de désorption Rd est la suivante
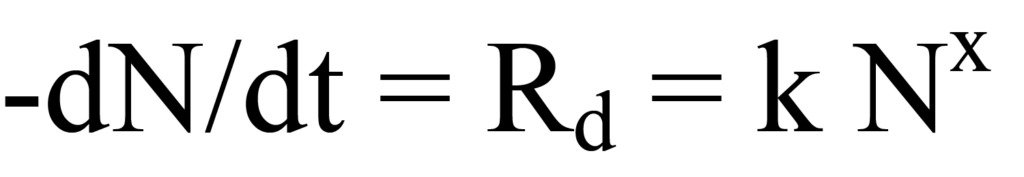
où k est la constante de vitesse, -N la concentration instantanée à la surface (nombre) de l'espèce adsorbée (négatif indiquant une diminution), et x l'ordre cinétique. Seul le cas le plus simple, la cinétique du premier ordre (non dissociative), est considéré ici ; par conséquent, x=1. Il convient de noter que le taux de désorption change constamment à mesure que le nombre de molécules restant à la surface diminue.
Les figures 8 et 9 illustrent le fait que l'énergie d'activation peut ou non être nécessaire pour que l'adsorption ait lieu. Cependant, ces figures montrent également qu'il existe toujours une barrière d'activation qui doit être surmontée pour que la molécule se désorbe, c'est-à-dire pour que la réaction de chimisorption de surface s'inverse. La relation entre la constante de vitesse et l'énergie dans les réactions dépendant de l'énergie telles que la désorption peut être exprimée sous la forme d'Arrhenius, à savoir
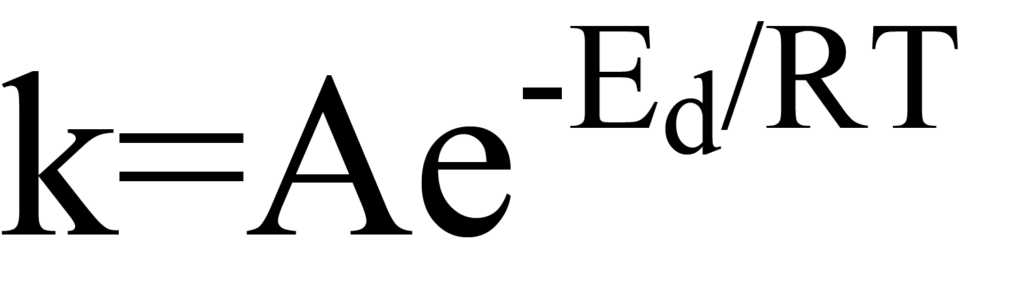
où Edes est l'énergie d'activation et l'indice "d" identifie le cas particulier de la désorption. Le facteur pré-exponentiel A peut être considéré comme le nombre de tentatives de la molécule par unité de temps pour s'échapper du puits de potentiel. T est la température (degrés Kelvin) et R la constante universelle des gaz. Le produit RT est l'énergie thermique, de sorte que l'exposant E/RT est le rapport entre l'énergie d'activation et l'énergie thermique. Si E est supérieur à RT, la probabilité de désorption est faible.
En substituant l'expression d'Arrhenius à l'équation 8, on obtient
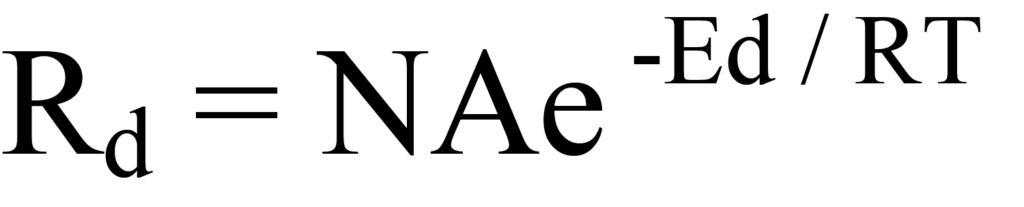
Il s'agit de l'équation de Polanyi-Wigner pour la vitesse de désorption du premier ordre. On peut expérimenter avec cette équation pour mieux comprendre ce qu'elle décrit en ce qui concerne la désorption. La figure 10 montre l'évolution du taux de désorption (Rd), de la couverture (N) et du rapport énergétique (Ed/RT) en fonction de la température, les valeurs étant normalisées sur l'axe vertical pour des raisons de commodité. Au cours d'une expérience réelle, la sortie du détecteur correspondrait au taux de désorption. La température Tm à laquelle le taux de désorption est maximal est facilement observable dans la figure 10, comme elle le serait sur un graphique du signal du détecteur (quantité d'espèces moléculaires évoluées) en fonction de la température.
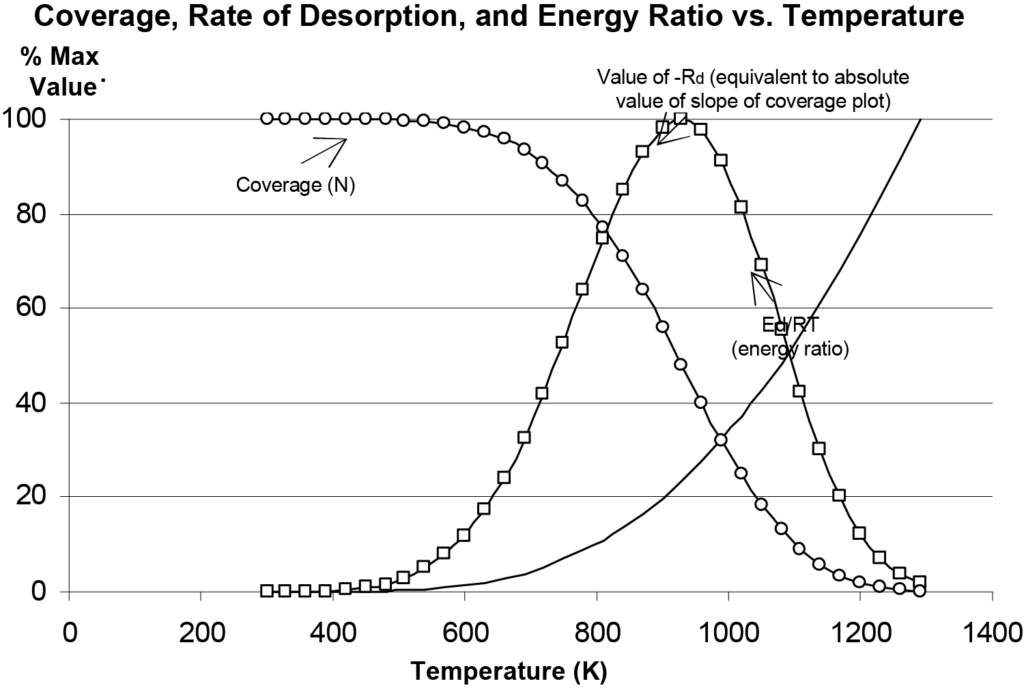
Note : La figure 10 a été produite par Microsoft Excel® en utilisant l'équation 14 et des valeurs arbitraires pour les paramètres. Cela permet d'expérimenter avec les paramètres et de comparer les différents tracés prédits par le modèle Polanyi-Wigner. On peut observer que les pics sont quelque peu asymétriques autour de Tm, l'augmentation de la valeur initiale de la couverture entraîne une augmentation de l'amplitude du pic, mais Tm reste constant. L'augmentation de la valeur de A entraîne le déplacement du pic vers des températures plus basses. L'augmentation de Ed entraîne l'élargissement du pic et la valeur maximale se produit à des températures plus élevées.
En extrayant le logarithme des deux côtés de l'équation 10, on obtient
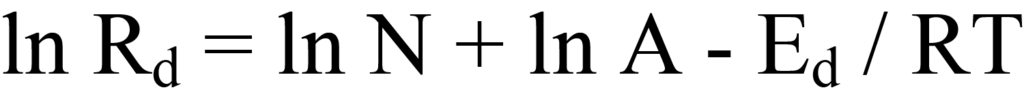
En regroupant cette équation sous forme linéaire, y = (m)x +(b), on obtient
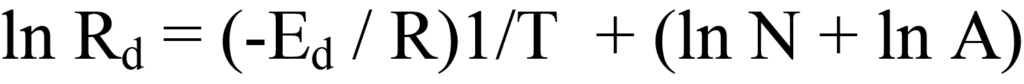
En traçant ln Rd en fonction de 1/T, on obtient une droite dont la pente est -Ed/R et l'ordonnée à l'origine (ln N + ln A). Le problème, ici, est que les valeurs numériques de Rd ne sont pas disponibles, de sorte que la dérivation doit être développée.
Au cours d'une analyse programmée en fonction de la température, l'échantillon est chauffé de façon linéaire, de sorte que la température T à tout moment t peut être calculée par la formule suivante
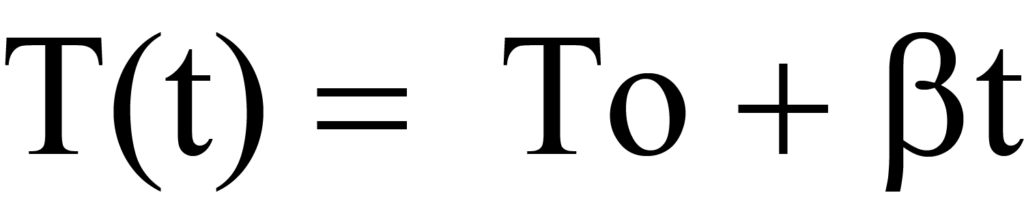
ou
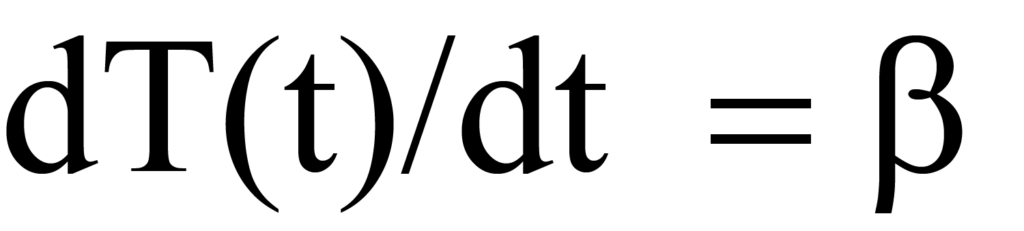
où β est la vitesse de chauffage ou la vitesse de rampe dT/dt en unités de degrés K par unité de temps. L'augmentation de la température entraîne une modification du nombre de molécules désorbées par unité de temps, exprimée par dN/dT. En multipliant la vitesse de chauffage en fonction du temps (dT/dt) en degrés/min par la vitesse de désorption en fonction de la température (-dN/dT) en molécules/degré), on obtient la vitesse de désorption en fonction du temps, Rd. Mathématiquement,
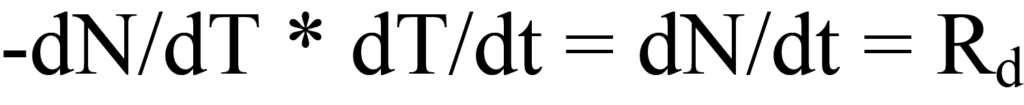
Le taux de chauffage β a été défini dans l'Eq. 15a comme dT/dt, par conséquent, la substitution dans le côté gauche de l'Eq. 12 donne
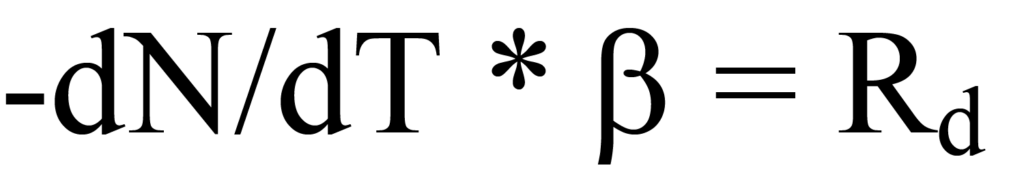
La substitution de l'Eq. 17 dans l'Eq. 14 donne
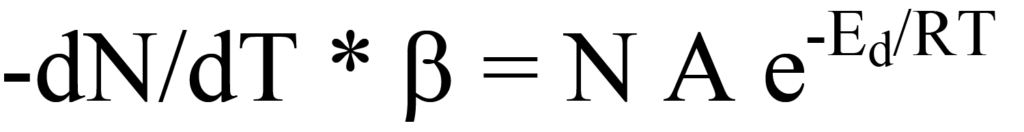
et le réarrangement donne le taux de désorption en fonction de la température,
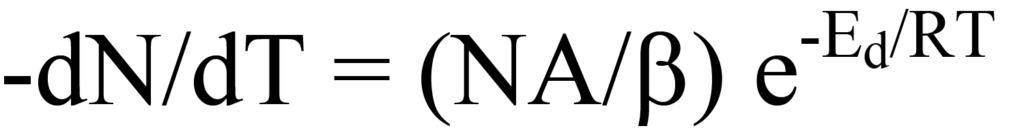
qui décrit le tracé de la vitesse de désorption de la figure 10. À mesure que la température augmente, la vitesse de désorption est maximale à Tm et, à cette température, la pente (dérivée première) de l'équation 15 est égale à zéro, c'est-à-dire,
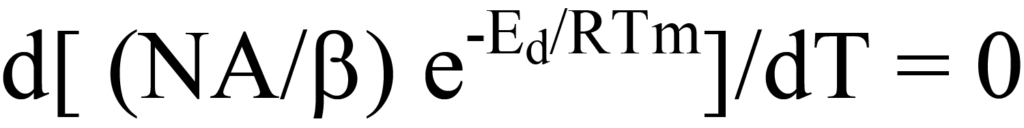
Pour des raisons de clarté et de continuité, nous rappelons au lecteur que l'équation 20 a la forme d(uv)/dT, où u = (A/b)N, v = e-Ed/RT, et N est une fonction de la température (T).
L'équation 20 donne
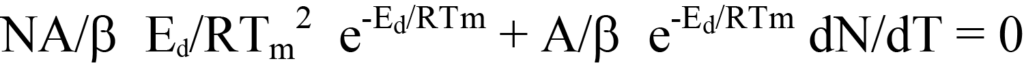
Maintenant, remplacez le côté droit de l'équation 19 par dN/dT (une valeur négative ; voir l'équation 12) dans l'équation 13 et faites le facteur. Cela donne
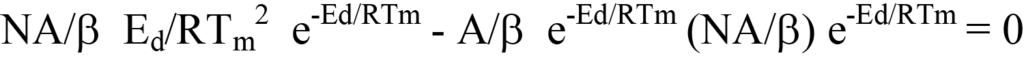
ou
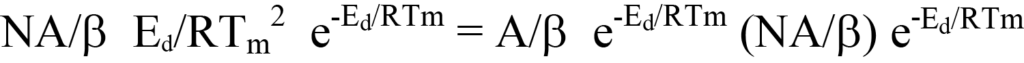
Si l'on élimine les termes communs aux côtés gauche et droit, il reste

Les valeurs de Tm, β et R sont connues ; A est inconnu ainsi queEd, la valeur recherchée. Une approche pour déterminer les inconnues consiste à exprimer les termes de l'équation 23 sous forme linéaire, y = mx + b. Cela peut se faire par réarrangement en
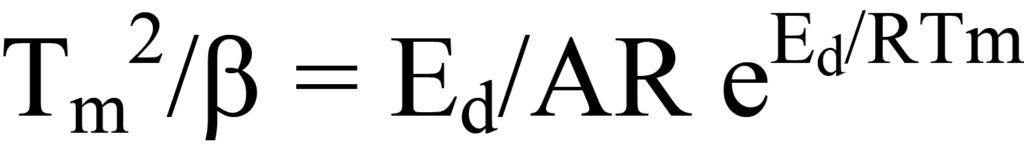
En prenant le logarithme de chaque côté, on obtient

une équation linéaire dans laquelle y = ln Tm 2/β, x = 1/Tm, m =Ed/R, et b = lnEd/AR. Le tracé de y en fonction de x pour une série de valeurs β permet de déterminerEd. Une autre forme de l'équation 25a que l'on trouve parfois dans la littérature consiste à développer les termes logarithmiques pour obtenir
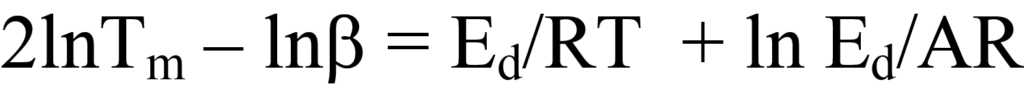
Une autre façon d'obtenir l'équation 25 à partir de l'équation 22a est de factoriser et de réarranger cette dernière équation, ce qui donne
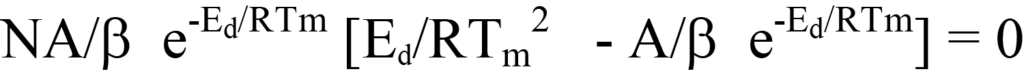
En divisant les deux côtés de cette équation par l'expression entre parenthèses, on obtient
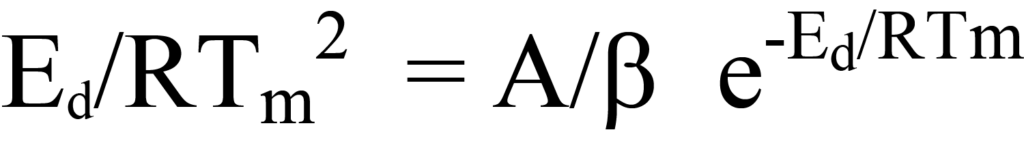
qui est identique à l'équation 23.
Il y a aussi le cas
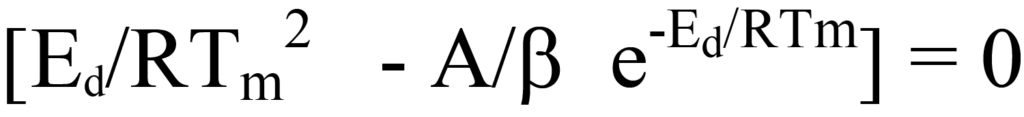
ou
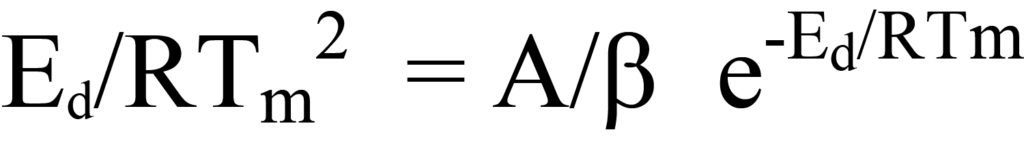
En divisant les deux côtés de l'équation 26 par les facteurs hors parenthèses ou en déduisant logiquement que, puisque ni le facteur NA/β ni e-Ed/RT n'a une valeur nulle pendant le processus de désorption, l'expression entre parenthèses de l'équation 26 doit être égale à zéro. En réarrangeant l'équation, on obtient
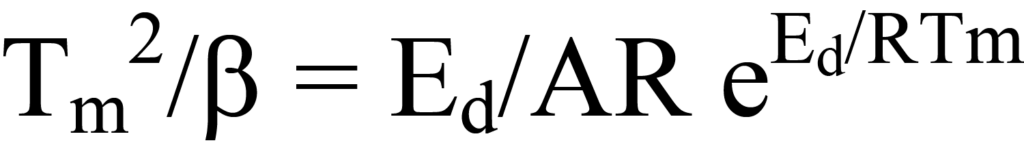
qui est identique à l'équation 24.
En suivant la méthode de Redhead, l'équation 25 est réarrangée comme suit :
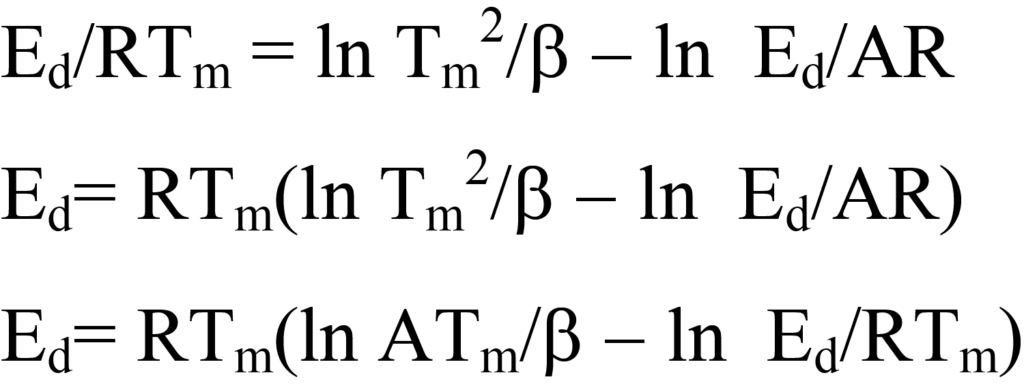
Cette équation est simplifiée en estimant le terme ln Ed/RTm du côté droit à 3,64, ce qui introduit une erreur de moins de 1,5 % pour des valeurs de A/β comprises entre108/K et1013/K, A étant typiquement approximé pour la cinétique du premier ordre à1013/s. Ainsi,
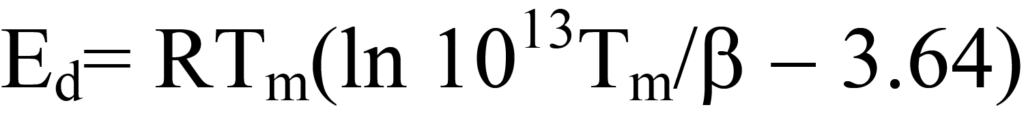
Tout comme la méthode du point unique pour déterminer la surface BET, cette méthode introduit une certaine erreur, mais elle permet d'estimerEd à partir d'un seul chromatogramme de désorption. (Schroeder et Gottfried). Plutôt que d'estimer la valeur de A, une série de chromatogrammes pourrait être obtenue pour différentes valeurs de β et les valeurs deEd et A déterminées à partir du tracé linéaire.
En utilisant la méthode de Redhead, il faut se rappeler qu'elle ne s'applique qu'à la cinétique du premier ordre. En ce qui concerne les réactions d'ordre supérieur, Stoltz avertit (Stoltze) : "Décrire E obtenu de cette manière comme une approximation est un euphémisme, complètement faux est une meilleure description".
Résumé
Les catalyseurs solides jouent un rôle "en coulisse" en rendant abordables et même possibles de nombreux avantages dont nous bénéficions dans la vie de tous les jours. La chimisorption est une étape fondamentale dans les réactions catalytiques, c'est pourquoi il est très utile de pouvoir étudier le processus de chimisorption à petite échelle en laboratoire. Les instruments analytiques modernes capables de caractériser ces réactions sont des outils puissants pour surveiller et contrôler la fabrication et l'application des catalyseurs et pour prédire les caractéristiques de performance des matériaux catalytiques nouvellement développés.
Ressources
James P. Olivier, Propriétés thermodynamiques des fluides confinés I : Experimental Measurements of Krypton Adsorbed by Mesoporous Silica From 80K to 130K, The 2nd Pacific Basin Conference on Adsorption Science and Technology, University of Queensland, Brisbane, Australia, May 2000.
Sven L.M. Schroeder et Michael Gottfried, Temperature- Programmed Desorption (TPD) and Thermal Desorption Spectroscopy (TDS), Freie University, Berlin (Internet http://userpage.chemie.fuberlin. de/~pcfp/V18/pdf/v18.pdf).
Paul L. Houston, Cornell University Chemical Kinematics and Reaction Dynamic, Ithaca.
Roger Nix, An Introduction to Surface Chemistry, Department of Chemistry, Queen Mary University of London (World Wide Web http:// www.chem.qmw.ac.uk/surfaces/scc/scat2_4.htm).
Simon J. Garrett, An Introduction to Surface Chemistry (lecture notes) , Michigan State University.
W.J. Moore, Physical Chemistry, Prentice-Hall, Inc (1972).
J.H. de Boer, The Dynamical Character of Adsorption, Oxford at the Clarendon Press (1953).
G.C. Bond, Heterogeneous Catalysis- Principles and Applications, Claredon Press, Oxford (1987).
John H. Sinfelt, Structure of Metal Catalysts, Review of Modern Physics, Vol. 51, No. 3 (1979).
Thomas H. Maugh, " Industry Steps Up Quest for Catalysts", High Technology, août 1984.
Scholten, Pijpers et Hustings, Surface Characterization of Supported and Nonsupported Hydrogenation Catalysts, Catal. Rev.-Sci. Eng., Vol. 27, No. 1, (1985).
A. Baiker, Méthodes expérimentales pour la caractérisation des catalyseurs. II. X-Ray Diffraction, Temperature- Programmed Desorption and Reduction, Thermogravimetry and Differential Thermoanalysis, International Chemical Engineering, Vol. 25, No. 1 (1985).
Philip Moriarty, Atoms and Molecules at Surfaces, School of Physics & Astronomy, University of Nottingham ( World Wide Web http:// www.nottingham.ac.uk/~ppzpjm/sect4_2.htm).
M. B. Raschke, U. Ho, Chemisorption energy of hydrogen on silicon surfaces, Physical Review B, Volume 63.
P. A. Redhead, The Ultimate Vacuum, Vacuum 12, 203-211, 1962.
Per Stoltze, Introduction to heterogeneous catalysis- Concepts and calculations, Department of Physics, Technical University of Denmark.
Gerard P. van der Laan, Kinetics, Selectivity and Scale Up of the Fischer-Tropsch Synthesis, thèse de doctorat, Université de Groningue, Pays-Bas, 1999.



