Resumen
La isoterma de adsorción química revela información sobre la superficie activa de un material y se ha empleado durante muchos años como herramienta analítica estándar para la evaluación de catalizadores. Las técnicas de reacción a temperatura programada han surgido a partir de los años 50 como un complemento indispensable de los análisis de isotermas de quimisorción en muchas áreas de la industria y la investigación. Este artículo ofrece una introducción a estas técnicas analíticas.
Introducción
El diseño óptimo y la utilización eficaz de los catalizadores requieren un conocimiento profundo de la estructura superficial y la química superficial del material catalítico. Los análisis de adsorción química (quimisorción) pueden proporcionar gran parte de la información necesaria para evaluar los materiales catalizadores en las fases de diseño y producción, así como tras un periodo de uso. El equipo analítico necesario puede ser relativamente barato, sencillo de manejar y rápido en comparación con otros equipos alternativos capaces de obtener la misma información.
Diferenciación entre adsorción física y química
Un material sólido suele presentar una distribución heterogénea de la energía superficial. Las moléculas de gas, vapor o líquido pueden unirse a la superficie si se acercan lo suficiente como para interactuar. Las discusiones de este artículo se limitan a la adsorción (y desorción) de gases o vapores en (o desde) superficies sólidas. El sólido se denomina adsorbente; la molécula de gas o vapor antes de ser adsorbida se denomina adsorbente y, mientras está unida a la superficie sólida, adsorbato.
La adsorción física es el resultado de una interacción sólido-gas relativamente débil. Se trata de una atracción física resultante de fuerzas de Van der Waal inespecíficas y relativamente débiles y de una energía de adsorción que no suele superar los 80 kJ/ mol, siendo las energías típicas considerablemente inferiores. Las moléculas adsorbidas físicamente pueden difundirse a lo largo de la superficie del adsorbente y normalmente no están ligadas a un lugar específico de la superficie. Al estar débilmente ligadas, la adsorción física se invierte fácilmente.
La adsorción también puede dar lugar a un complejo superficial, una unión mucho más fuerte que un enlace físico con calores de adsorción de hasta unos 600 kJ/mol para enlaces C-N y 800 kJ/mol para enlaces químicos. Un enlace químico implica el intercambio de electrones entre el adsorbato y el adsorbente y puede considerarse como la formación de un compuesto superficial. Debido a la fuerza del enlace, la adsorción química es difícil de revertir.
La adsorción física tiene lugar en todas las superficies siempre que las condiciones de temperatura y presión sean favorables. La quimisorción, sin embargo, es altamente selectiva y sólo se produce entre determinadas especies adsorbentes y adsorbidas y sólo si la superficie químicamente activa se limpia de moléculas adsorbidas previamente.
En condiciones adecuadas, la adsorción física puede dar lugar a que las moléculas adsorbidas formen múltiples capas. La quimisorción, en el caso típico, sólo procede mientras el adsorbente pueda entrar en contacto directo con la superficie; es, por tanto, un proceso de una sola capa. Puede haber excepciones si el adsorbente es muy polar, como por ejemplo el NH3. La adsorción física y química puede producirse en la superficie al mismo tiempo; una capa de moléculas puede adsorberse físicamente sobre una capa quimisorbida subyacente. La misma superficie puede presentar adsorción física a una temperatura y adsorción química a una temperatura superior. Por ejemplo, a la temperatura del nitrógeno líquido (77 K), el gas nitrógeno se adsorbe físicamente en el hierro, pero a 800 K, un nivel de energía demasiado alto para los enlaces de adsorción física, el nitrógeno se adsorbe químicamente para formar nitruro de hierro (Moore).
Aplicaciones generales de la sorción de gases como herramienta analítica
A partir de las observaciones del proceso de adsorción pueden determinarse diversas características de la superficie. La cantidad de moléculas absorbidas por una superficie depende de diversas variables, como la temperatura, la presión, la distribución de la energía superficial y la superficie y porosidad del sólido. La relación entre la cantidad de moléculas adsorbidas y la presión a temperatura constante se denomina isoterma de adsorción.
Las isotermas físicas de adsorción y desorción son importantes para caracterizar la superficie en su conjunto. El más mínimo cambio en la forma de la isoterma trazada es indicativo de una característica particular de la superficie. Los análisis de los datos de la isoterma de adsorción física revelan el área total de la superficie, el volumen y el área de los mesoporos y microporos, el volumen total de los poros, la distribución del volumen y el área de los poros por tamaño de poro y la distribución de la energía superficial. Así pues, la adsorción física es una herramienta importante en el estudio de los catalizadores, sobre todo para evaluar la estructura del soporte.
La isoterma de adsorción química también evalúa la superficie, pero es selectiva en el sentido de que sólo sondea las zonas activas, es decir, aquellas capaces de formar un enlace químico con el gas o vapor adsorbente. Esta selectividad afecta tanto a la molécula de la sonda como a la composición molecular o atómica del material activo, por lo que la elección de las moléculas de la sonda es mucho más importante en los ensayos de quimisorción. Aunque los ensayos de quimisorción isotérmica son importantes para caracterizar las superficies activas, los ensayos de temperatura programada lo son aún más. La forma básica de los datos producidos por estas pruebas se asemeja a un cromatograma de temperatura frente a la cantidad desorbida.
Catalizador Básico
Un catalizador influye en la velocidad de una reacción química. La palabra "velocidad" es la más importante en esta descripción porque un catalizador no puede inducir una reacción que no esté permitida por las leyes de la termodinámica. Un catalizador sólo puede aumentar la velocidad a la que la reacción se aproxima al equilibrio.
Los catalizadores heterogéneos incluyen metales, óxidos metálicos y ácidos sólidos. Los metales puros pueden emplearse como catalizadores sólidos o dispersarse en forma de pequeños granos en la superficie de un material de soporte como TiO2, ZrO2, Al2O3 o SiO2. La preparación de un catalizador soportado implica seleccionar precursores de los componentes activos y cualquier promotor necesario y mezclarlos en un disolvente. La mezcla forma un precipitante, o se utiliza para recubrir un soporte inerte, o se utiliza para impregnar un soporte. Finalmente, el metal activo o precursor se dispersa en el soporte. El producto se seca, se mezcla con un aglutinante o un agente de formación y, a continuación, se tritura, se granula, se extruye o se le da cualquier otra forma. Por último, el material se calcina y se activa por oxidación, reducción u otros medios.
Un tipo de catalizador sin soporte está compuesto de metal puro. Los catalizadores de metal Raney, por ejemplo, se preparan disolviendo una aleación de aluminio y níquel en una solución de hidróxido de sodio, que disuelve el componente de aluminio. El producto final es una "esponja" metálica activa muy porosa, de la que se han eliminado todos los demás materiales de andamiaje. Otro catalizador metálico sin soporte se produce fusionando o sinterizando óxidos metálicos con promotores, formando así una red de poros en toda la masa metálica (van der Laan). Un ejemplo de ello es un catalizador de síntesis de amoníaco de hierro fundido.
Los catalizadores zeolíticos forman otro grupo. Las zeolitas son aluminosilicatos hidratados y se utilizan ampliamente en la industria química y en operaciones de refinado. Su actividad depende de la proporción entre sílice y alúmina. Los catalizadores amorfos de sílice-alúmina tienen una actividad menor que los catalizadores zeolíticos y se aplican en operaciones de hidrocraqueo suave. Se requiere una superficie ácida para el craqueo y la acidez está asociada a los átomos de oxígeno unidos a los átomos de aluminio.
Quimisorción y catalizadores
El proceso catalítico puede generalizarse mediante una breve secuencia de pasos como la siguiente. Consideremos las moléculas reactivas A y B en la masa gaseosa sobre una superficie sólida que soporta un conjunto de sitios metálicos activos S. Si la molécula A se adsorbe químicamente en uno de los sitios activos, se forma un complejo de superficie A. A continuación, A reacciona con B formando la molécula A+B, que escapa del sitio S y se regenera. A continuación, A reacciona con B formando la molécula A+B, que escapa del sitio, regenerando así el sitio S.
La adsorción de al menos una de las moléculas reactivas es necesaria para que se produzca la catálisis. Si la aceleración de la velocidad de reacción se debiera simplemente a la concentración de moléculas en la superficie, la catálisis sería el resultado de la adsorción física de los reactivos. La quimisorción es un paso esencial, la molécula adsorbida forma un complejo superficial intermedio que es más receptivo a la reacción química. La dependencia de la catálisis de la quimisorción es una de las razones por las que la quimisorción es una técnica analítica tan informativa en el estudio de la catálisis: la química que se produce en la aplicación del catalizador se observa directamente en el laboratorio.
El rendimiento de un catalizador depende de varias variables. En primer lugar, los lugares de adsorción deben ser numerosos y estar disponibles para las moléculas reactivas. En algunos casos, los granos de metal activo están en la superficie, pero también hay granos situados bajo la superficie y no disponibles para los reactivos. La simple ubicación de los puntos de adsorción en la superficie no basta para garantizar un rendimiento óptimo. Por ejemplo, algunos sitios potenciales de adsorción pueden estar situados en la profundidad de un microporo que es demasiado estrecho para que entre la molécula reactiva o para que salga el producto de reacción; en este caso, el sitio de la superficie no puede ser un participante activo en la quimisorción. Un sitio podría estar situado a lo largo de un camino tortuoso que impida el flujo eficiente de reactivos hacia el sitio activo y de productos fuera del sitio. Dado que muchos metales activos son muy caros, un criterio de diseño importante es maximizar el número de sitios activos por unidad de metal.
La actividad catalítica depende de la rapidez con la que se produce la quimisorción y de la fuerza (energía) del enlace de quimisorción. Si el enlace es demasiado débil, la molécula puede desorberse antes de reaccionar; si es demasiado fuerte, puede retrasarse la liberación del producto y la regeneración del sitio. Para estudiar la distribución de la energía superficial pueden utilizarse métodos de quimisorción isotérmica y métodos de quimisorción a temperatura programada.
La quimisorción descrita anteriormente se refiere al uso de catalizadores en diversas aplicaciones. Las mismas reacciones, sólo que a menor escala, también pueden producirse en un tubo de muestra en condiciones controladas que permitan estudiar el proceso. Se trata de la quimisorción utilizada como técnica analítica. Los análisis de quimisorción se aplican para caracterizar físicamente un material catalizador, para determinar la eficiencia relativa de un catalizador en la promoción de una reacción particular, para estudiar el envenenamiento del catalizador y en el seguimiento de la degradación de la actividad catalítica con el tiempo de uso. Entre las diversas técnicas instrumentales utilizadas en el análisis de catalizadores, las técnicas de quimisorción son las más universalmente empleadas.
Instrumentos de quimisorción
Los análisis de quimisorción isotérmica se obtienen mediante dos técnicas de quimisorción: a) quimisorción volumétrica estática, y b) quimisorción dinámica (gas que fluye).
La técnica volumétrica es conveniente para obtener una medición de alta resolución de la isoterma de quimisorción desde una presión muy baja hasta la presión atmosférica, esencialmente a cualquier temperatura, desde casi ambiente hasta 1000+ oC. Las realizaciones comerciales de esta técnica están casi exclusivamente automatizadas. La obtención de una isoterma de alta resolución requiere muchos pasos de dosificación precisos en busca del punto de equilibrio, y muchos pasos de presión que, sin automatización, serían un procedimiento lento y propenso a errores.
La técnica de flujo de gas (dinámica) funciona a presión ambiente. Una vez limpia la muestra, se administran pequeñas inyecciones (dosis) de cantidades exactamente conocidas de adsorbente en pulsos hasta que la muestra se satura, de ahí el nombre de "quimisorción en pulsos". Un detector de conductividad térmica calibrado (TCD) controla la cantidad de adsorbente que no es absorbida por los metales activos. Esta cantidad se resta de la cantidad inyectada para obtener la cantidad adsorbida en cada inyección. Estas cantidades se suman para determinar la capacidad de la masa de la muestra. Las inyecciones pueden realizarse mediante jeringa o mediante una válvula de inyección de bucle manual o automatizada de tipo HPLC. Dado que el punto final (saturación) es el único punto medido, el número de inyecciones puede ser reducido y no se producen cambios de presión. Así pues, la técnica de flujo de gas suele realizarse manualmente, aunque también existen instrumentos automatizados.
En las últimas décadas, los analizadores de quimisorción de temperatura programada se han convertido en herramientas muy valiosas para la caracterización de catalizadores. El método se denomina a veces simplemente reacciones programadas por temperatura, e incluye la desorción programada por temperatura (TPD), la reducción programada por temperatura (TPR) y la oxidación programada por temperatura (TPO). El método de quimisorción dinámica es especialmente adecuado para realizar análisis de programas de temperatura.
Análisis de quimisorción
La versatilidad de la técnica de quimisorción queda ilustrada por la cantidad de información sobre un material que puede obtenerse. Parte de esta versatilidad se ha puesto de manifiesto en el análisis anterior. En las secciones siguientes se exploran otras posibilidades.
En los ejemplos que siguen, sólo se consideran catalizadores compuestos por una única especie activa para simplificar los cálculos. En el caso de catalizadores metálicos mixtos, muchas de las ecuaciones que se presentan a continuación requerirían sumar una serie de términos, uno por cada especie adsorbente y cada término ponderado por la contribución fraccional de la especie al conjunto.
La isoterma de quimisorción
Una isoterma es un conjunto de puntos de datos de cantidad adsorbida frente a presión que caracterizan el proceso de adsorción a una temperatura constante. Para entenderlo mejor, consideremos un reactor de volumen constante V que contiene una muestra y una cantidad de moléculas de gas N. La teoría de la quimisorción supone que la superficie activa de un sólido contiene un número fijo Ns de sitios de adsorción y que sólo una molécula a la vez puede ocupar un sitio. Las moléculas en el tubo de muestra que compiten por un sitio de adsorción representan una concentración C que puede expresarse en general resolviendo la ley de los gases , o
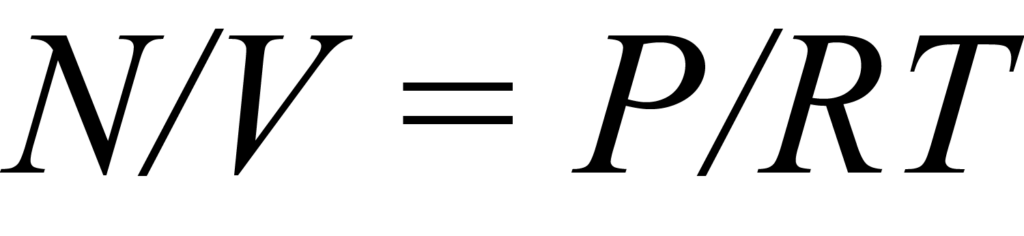
donde N y V se han definido previamente, P es la presión, T la temperatura y R la constante de los gases.
Para un sistema gas-sólido específico en equilibrio bajo condiciones específicas de temperatura y presión, una cierta fracción ϑ del número total de sitios disponibles Ns será ocupada por nads moléculas adsorbidas. Esto se describe mediante
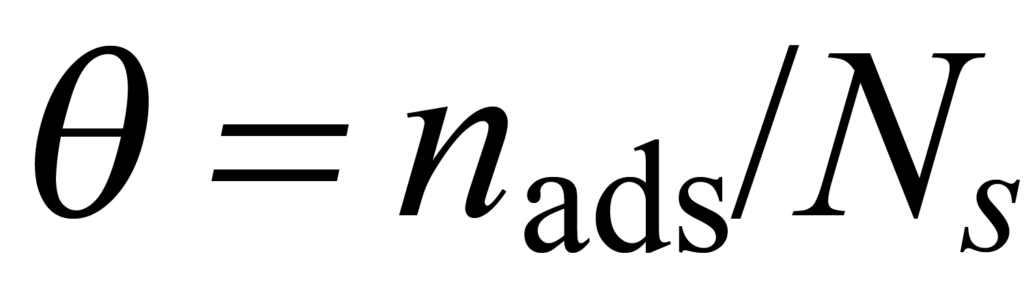
La velocidad a la que se alcanza el equilibrio depende de la concentración de las moléculas implicadas, así como de otras consideraciones, estas últimas agrupadas en un parámetro denominado "constante de velocidad" k.
Para el proceso de adsorción, la concentración de moléculas no adsorbidas que compiten por los sitios restantes es C-ϑC y la velocidad de adsorción es kads(C-ϑC) o kadsC(1-ϑ). Para la desorción, la fracción de moléculas desorbidas es ? y la velocidad de desorción es kdesϑ. Cuando se alcanza el equilibrio, la velocidad de adsorción y desorción son iguales, es decir,
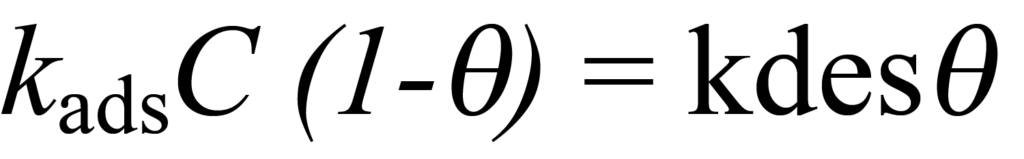
o
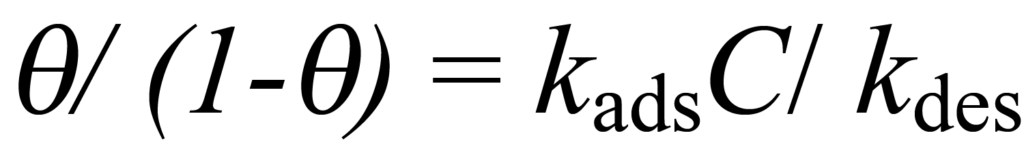
Utilizando K para representar kads/kdes y resolviendo para ϑ se obtiene
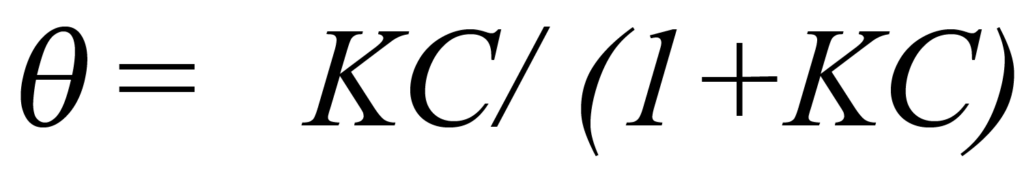
Recordando que el ambiente es de volumen constante V y temperatura constante T, cambiar la presión es la única forma que queda para cambiar la concentración C. La Ec. 1 establece que la concentración es directamente proporcional a la presión, por lo tanto si la constante de proporcionalidad concentración-presión se combina con K y se representa por b, la Ec.4 puede escribirse
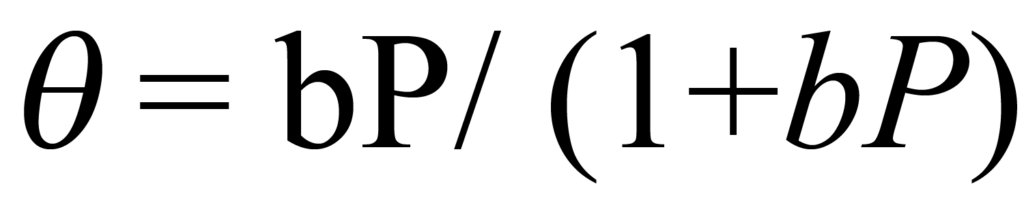
Se trata de la famosa isoterma de Langmuir, que describe con precisión la mayoría de las isotermas de quimisorción que no implican fragmentación (disociación) de la molécula adsorbente. La isoterma es asintótica a ϑ = 1, que es la cobertura total, o terminación de la monocapa.
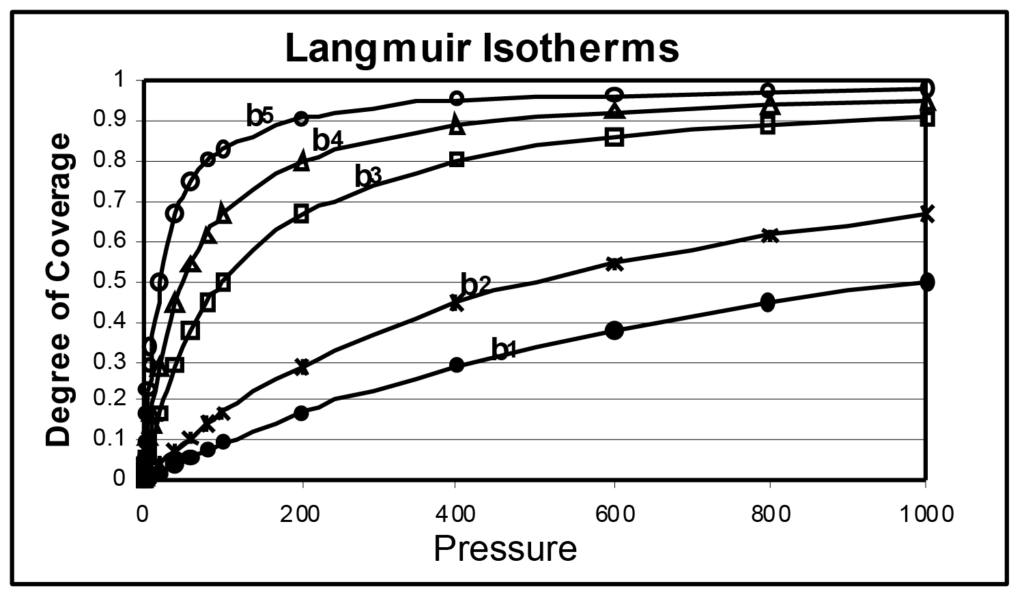
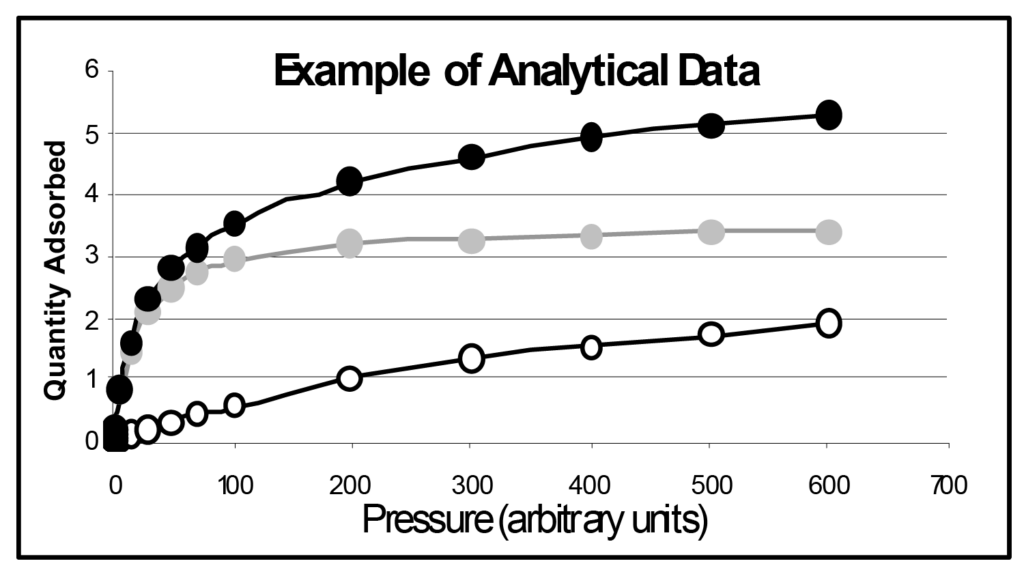
En la figura 1 se ilustran varias isotermas de Langmuir, cuya única diferencia es la magnitud de b. El parámetro b está directamente relacionado con la energía superficial; al aumentar ésta, aumenta la probabilidad de adsorción a una presión dada. El parámetro b está inversamente relacionado con la temperatura, que, al aumentar, incrementa la energía molecular y disminuye la probabilidad de adsorción a una presión dada.
En la ilustración, b1 es menor que b2, que es menor que b3, y así sucesivamente, siendo b5 50 veces mayor que b1. De ésta puede deducirse que la misma muestra analizada a cinco temperaturas diferentes produciría un conjunto similar de isotermas, al igual que cinco muestras diferentes de distinta energía superficial medidas a la misma temperatura de análisis.
La obtención de una isoterma de quimisorción no es necesariamente una medición directa, como se ilustra en las Figuras 2. En el caso típico indicado por estas ilustraciones, habrá moléculas (indicadas por R) débilmente adsorbidas a la superficie del soporte y también habrá una monocapa quimisorbida de moléculas (indicada por I) sobre la superficie activa. Dependiendo de la presión y la temperatura, también puede haber moléculas adsorbidas físicamente sobre la monocapa quimisorbida.
La adsorción débil se denomina adsorción reversible y la quimisorción fuerte, irreversible, de ahí las etiquetas R e I de la ilustración. La isoterma inicial será una combinación de adsorción reversible e irreversible.
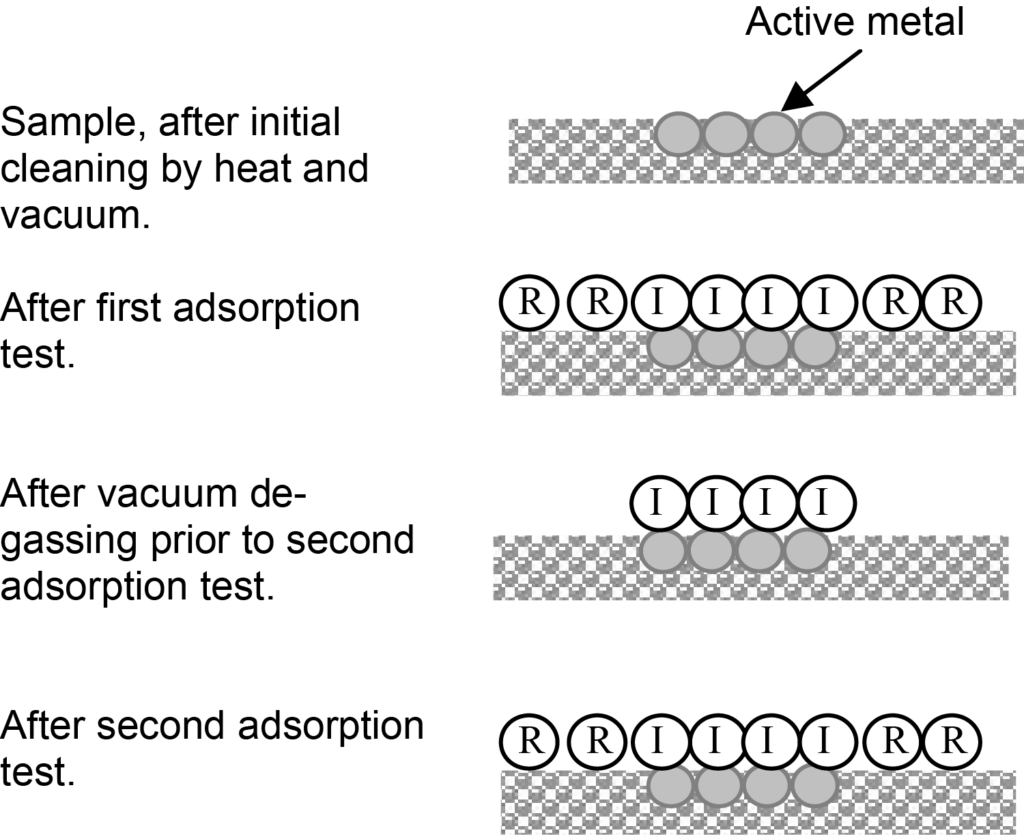
Las contribuciones reversibles e irreversibles a una isoterma combinada pueden distinguirse realizando un segundo ensayo de adsorción. Tras la prueba de adsorción inicial y antes de la segunda prueba, la muestra se somete únicamente a vacío, lo que provoca la desorción de las moléculas débilmente adsorbidas y deja únicamente las moléculas que han formado un enlace de quimisorción fuerte con la superficie activa.
El segundo ensayo de adsorción se realiza en las mismas condiciones que el ensayo inicial, pero esta vez la superficie activa ya está cubierta por una monocapa quimisorbida. La absorción de adsorbato será sólo la asociada a la adsorción reversible. Restando la cantidad adsorbida reversiblemente de la isoterma combinada en cada valor de presión se obtiene una isoterma de adsorción ireversible. Los resultados analíticos se ilustran en la figura 3.
Número de sitios activos accesibles
Tanto las técnicas volumétricas estáticas como las dinámicas de quimisorción pueden utilizarse para medir la cantidad de gas necesaria para formar una monocapa de quimisorbato en una superficie activa. El método dinámico sólo produce un único punto de datos, que corresponde a la cantidad de adsorbato necesaria para saturar la superficie activa con una monocapa. La capacidad de monocapa de la superficie activa se calcula a partir de la isoterma de adsorción irreversible derivada que se ilustra en la figura 3. Dado que la isoterma de adsorción irreversible es asintótica al valor de la cobertura total (de la superficie activa), la meseta de la isoterma se extrapola al eje y para obtener el valor de la capacidad monocapa. La extrapolación al eje y de la sección lineal de las isotermas reversibles-irreversibles combinadas también proporciona un valor razonable para la cantidad de cobertura de la monocapa.
El método de quimisorción dinámica requiere la inyección (manual o automática) de pequeñas cantidades, conocidas con precisión, de adsorbente en una corriente de gas portador inerte que fluye a través del lecho de la muestra.
La presión parcial del adsorbente aumenta momentáneamente sobre la muestra a medida que pasa el pulso y se adsorbe una parte o la totalidad de la cantidad de gas inyectada. El gas no adsorbido es barrido a través del detector, donde queda registrado. Nótese que, a diferencia del método estático (descrito anteriormente), la adsorción neta es sólo adsorción irreversible porque, después de que pasa el pulso de adsorbente, la presión parcial del adsorbente en el gas a granel es esencialmente cero y las moléculas débilmente adsorbidas que escapan son barridas hacia el detector por la corriente portadora.
A medida que se forma la monocapa en la superficie activa, la muestra absorbe cada vez menos adsorbente inyectado. Las inyecciones continúan hasta que la muestra se satura, como se indica cuando todos los picos subsiguientes del detector mantienen el mismo tamaño. Los resultados de una prueba de este tipo se muestran en la figura 4, que ilustra la salida del detector. El área de cada pico es proporcional a la cantidad de la inyección que no se adsorbió.
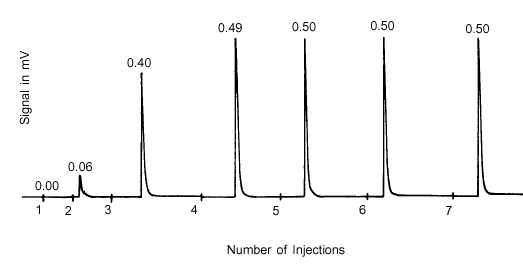
En la figura 4, el área integrada de los picos asociados a las inyecciones 5, 6 y 7 indica que no se produjo adsorción, por lo que el área activa está saturada. Toda la primera inyección fue adsorbida, al igual que el 88% de la inyección 2, el 20% de la inyección 4 y el 2% de la inyección 3. A partir de la cantidad conocida de cada inyección, la fracción de cada inyección que fue adsorbida y la masa del material de muestra, se determina el número de moles de gas adsorbente absorbidos por cada gramo de muestra.
El proceso de adsorción puede implicar la separación de un adsorbente molecular en dos o más entidades moleculares o atómicas (iones o radicales), formando cada entidad un enlace con sitios activos individuales de la superficie. Esto se denomina adsorción disociativa o de segundo orden, en contraste con la adsorción de primer orden o no disociativa, en la que la molécula adsorbente permanece intacta durante la adsorción. El resultado de la adsorción disociativa es que el número de moléculas adsorbentes Nm que se ha determinado que han sido absorbidas por gramo de muestra no es igual al número de átomos de superficie activa que participaron en la quimisorción.
Para determinar el número de átomos superficiales implicados, hay que tener en cuenta la estequiometría de la reacción superficial. La estequiometría se refiere a la relación entre las cantidades de sustancias que reaccionan juntas en una reacción química concreta y las cantidades de productos que se forman. Por ejemplo, una molécula de hidrógeno (H2) puede disociarse en dos átomos de hidrógeno y reaccionar con dos átomos de superficie activa, como es el caso del Pt. Así, el número de moléculas adsorbidas por la superficie activa debe multiplicarse por un factor estequiométrico Fs (donde Fs = 2 en el caso del ejemplo) para llegar al número de átomos de superficie Ns o "sitios". Matemáticamente,
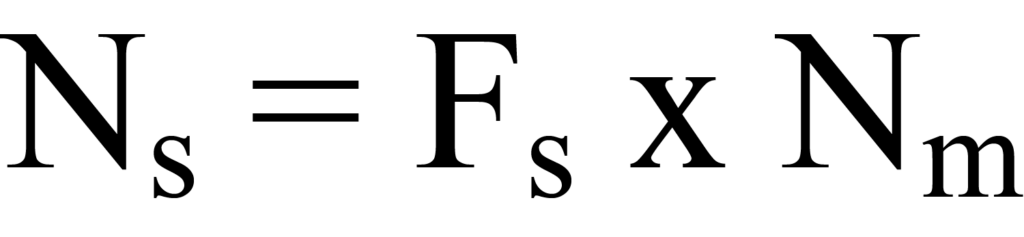
donde Ns y Nm se determinan por gramo de muestra.
La molécula adsorbente puede unirse a la superficie activa en más de una configuración. Debe tenerse en cuenta la proporción de moléculas que se unen de cada forma posible, lo que a menudo da lugar a un factor de estequiometría no entero.
Superficie activa
La superficie activa específica se obtiene a partir del número de moléculas Ns adsorbidas en la superficie activa de un gramo de muestra. La superficie activa específicaAA se determina multiplicando el áreaAm ocupada por una molécula de la superficie por el número de moléculas adsorbidas por gramo Ns. El área ocupada por un solo átomo procedente de la adsorción puede localizarse normalmente en la bibliografía. También puede determinarse experimentalmente, si es necesario, determinando el área superficial BET de una muestra de material activo puro utilizando N2, determinando a continuación la absorción molar del adsorbente activo en la misma muestra.
Dispersión y porcentaje de metal
La dispersión de metal describe la relación entre el número de átomos de metal activo disponibles para la reacción y el número total de átomos de metal en el material del catalizador. La cantidad de metal activo incorporado por unidad de masa de material de soporte está disponible a partir de la fórmula de fabricación; esto dará como resultado la cantidad de átomos de metal activoNT por unidad de masa de catalizador. El análisis de quimisorción descrito anteriormente se utiliza para determinar la cantidad de metal activo por gramo disponible para la reacción. El porcentaje de dispersión es la relación entre la cantidad disponible y la cantidad total de moléculas activas multiplicada por 100%, es decir
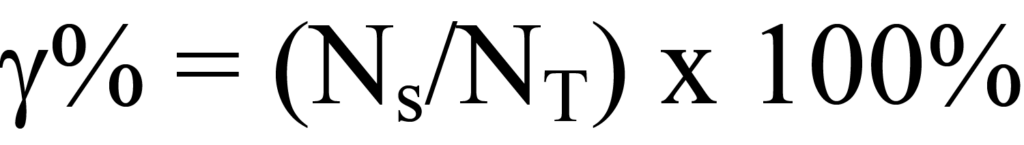
La dispersión se expresa como una relación entre el metal disponible y el metal total. Otra fracción que se determina a partir del procedimiento de fabricación es la relación entre el peso del metal y la masa del material a granel del catalizador, expresada como fracción decimal o como porcentaje.
Tamaño de las partículas activas (cristalitos)
Esta estimación del tamaño de la partícula activa es un cálculo geométrico basado en la suposición de que la forma del cristalito es de geometría regular, siendo normalmente una esfera la geometría elegida. Lo que se conoce a partir de cálculos anteriores es la superficie activa por gramo de material de muestraAA y, a partir del procedimiento de fabricación del catalizador, la proporción fraccionaria de metal en la masa del catalizador.
El cálculo emplea una expresión de la geometría del grano. A partir de la geometría regular supuesta, el diámetro puede expresarse en términos de área y volumen. El volumen de metal activo no se conoce, aunque sí la densidad atómica o molecular ρm; por tanto, el volumen puede expresarse en términos de densidad. El área de metal activo por gramo de muestra se midió previamente, proporcionando un valor para el área por unidad de masa de metalAm (m2/g). Sustituyendo estas expresiones en la relación general para D = f(A,V) se obtiene
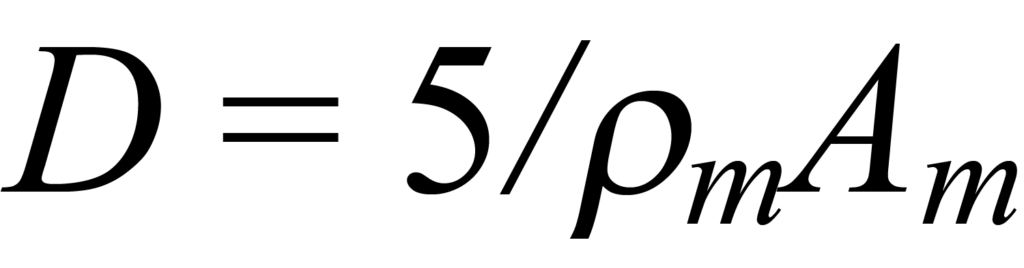
para un grano cúbico en la superficie (cinco de seis caras expuestas) y
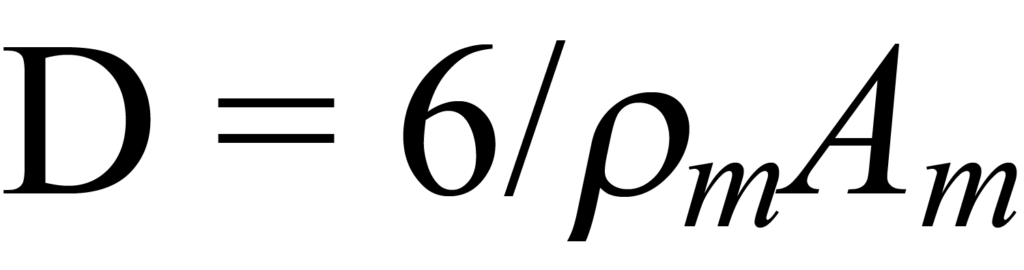
para una partícula de forma semiesférica.
El diámetro calculado mediante la ecuación anterior representa el diámetro medio de los granos de metal activo sobre los que se ha producido la adsorción.
Quimisorción a temperatura programada
Como su nombre indica, la quimisorción programada por temperatura es un método que implica los efectos de la temperatura en las reacciones superficiales. Hay tres reacciones principales que se estudian en función de la temperatura: 1) desorción, 2) reducción y 3) oxidación.
La desorción programada por temperatura mediante el método dinámico consiste en colocar una muestra en una célula de muestra y pretratarla para eliminar cualquier especie adsorbida de la superficie activa. A continuación, un gas o vapor seleccionado se adsorbe químicamente en los sitios activos hasta que se alcanza la saturación, tras lo cual las moléculas adsorbidas restantes se eliminan con un gas inerte. La temperatura (energía) aumenta a un ritmo controlado mientras se mantiene un flujo constante de gas inerte sobre la muestra. El gas inerte y las moléculas desorbidas se controlan mediante un TCD. La señal del TCD es proporcional a la cantidad de moléculas desorbidas a medida que la energía térmica, controlada por un termopar, supera la energía de enlace. Las cantidades desorbidas a temperaturas específicas proporcionan información sobre el número, la fuerza y la heterogeneidad de los sitios de adsorción. Los datos de análisis suelen representarse como la cantidad desorbida en función de la temperatura, o como la temperatura y la cantidad desorbida representadas en función del tiempo.
La figura 5 es un perfil TPD típico de desorción de hidrógeno a partir de Pt soportado sobre Al2O3. El primer pico se obtiene a una temperatura de 393 K. Este pico corresponde a la débil adsorción de hidrógeno y puede estar relacionado con la desorción del soporte o con una débil quimisorción. El segundo pico obtenido a la temperatura inmediatamente superior (493 K) corresponde probablemente al desbordamiento de hidrógeno debido a la presencia de Pt sobre la alúmina. El tercer y último pico obtenido a 570 K corresponde al hidrógeno quimisorbido por el Pt. Este pico se cuantifica para estimar la cantidad de Pt disponible para la actividad catalítica en un reactor. Las energías de enlace pueden cuantificarse mediante el método TPD, como se verá más adelante.

La reducción programada por temperatura es un método por el cual una mezcla de gas reductor, como el hidrógeno diluido en un gas inerte, fluye sobre una muestra de un óxido. La temperatura inicial suele ser inferior a la temperatura de reducción. A continuación, se aumenta la temperatura de la muestra a un ritmo constante y, a medida que comienza la reducción, se consume el hidrógeno de la mezcla portadora, lo que se detecta mediante un TCD. Cuando cesa la reducción, no se consume más hidrógeno y la conductividad térmica del gas del tubo de muestra vuelve a la línea de base. Pueden detectarse varios picos de reducción a lo largo de la rampa de temperatura, ya que la reducción probablemente se iniciará a varios niveles de energía térmica. Cada pico corresponde entonces a un óxido diferente y la amplitud de cada pico es proporcional a la velocidad de reacción.
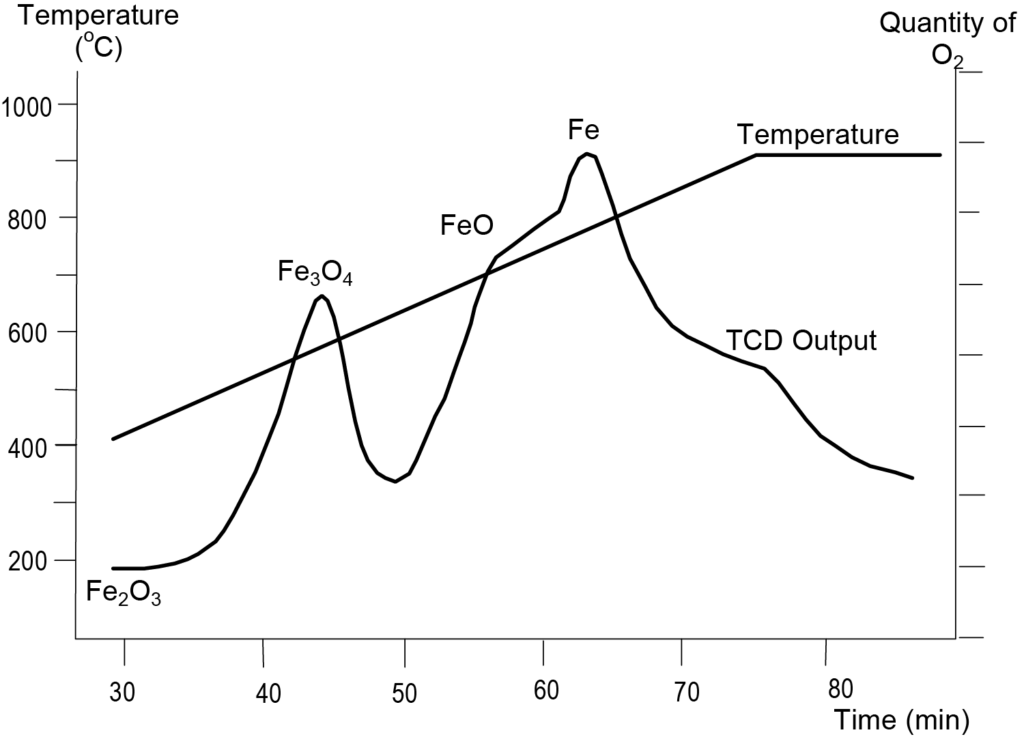
El óxido de hierro en forma de hematites presenta tres fases de reducción correspondientes a los tres óxidos de hierro. Se realizó un análisis TPR de la hematites utilizando un gas reductor compuesto por un 10% de hidrógeno en argón y una velocidad de rampa de temperatura de 10 °C/min. La figura 6 muestra los resultados. El primer pico de reducción aparece a 575 K, correspondiente a la transición de Fe2O3 a Fe3O4. El pico a 627 K corresponde a la transformación de Fe3O4 en FeO. El pico final a 748 K corresponde a la transición de FeO a Fe. Las posiciones de los máximos mostrados en la Figura 6 pueden diferir algo de una muestra a otra en función del tamaño de las partículas de Fe2O3y de otros parámetros como la velocidad de rampa de la temperatura.
La oxidación programada a temperatura puede utilizarse para determinar la cantidad de especies reducidas (también denominada grado de reducción), pero la dificultad de este tipo de análisis estriba en alcanzar la oxidación total. Más comúnmente, la TPO se utiliza en aplicaciones como el estudio de la cinética de la coquización, la evaluación de la quema de carbono del catalizador, la determinación de las diferentes formas de depósitos carbonosos presentes en los catalizadores tras una reacción de descomposición de CO o, dicho de forma más general, las mediciones del consumo de oxígeno y los rendimientos de los productos.
La muestra se calienta a un ritmo uniforme a medida que el gas reactivo, normalmente entre un 2% y un 5% de oxígeno, se aplica a la muestra en pulsos o, alternativamente, como un flujo constante. La reacción de oxidación se produce a una temperatura específica que da lugar a una absorción de oxígeno. La cantidad de oxígeno consumido durante la reacción está relacionada con la cantidad de una especie en la superficie.
Energía superficial y cinética de primer orden
Los mecanismos de adsorción y desorción se componen de alguna combinación de pasos cinéticos elementales. Por ejemplo, una molécula diatómica puede adsorberse o, al acercarse a la superficie, puede fragmentarse en átomos que se adsorben independientemente. El primer caso se denomina quimisorción no disociativa o cinética de primer orden; el segundo, quimisorción disociativa o cinética de segundo orden. La cinética de primer orden será el tema principal de las siguientes discusiones.
La interacción de una molécula aislada con una superficie puede representarse en un gráfico de energía potencial en función de la distancia a la superficie, como se muestra en la figura 7. A modo de comparación, la Figura 7 también incluye un diagrama de energía para la fisisorción, así como la ruta de energía neta tomada por la molécula quimisorbida. Al examinar este diagrama, debe entenderse que se trata de una representación básica de una molécula que se acerca a una superficie y que sólo tiene en cuenta la energía en función de la distancia a la superficie, sin tener en cuenta otras variables.
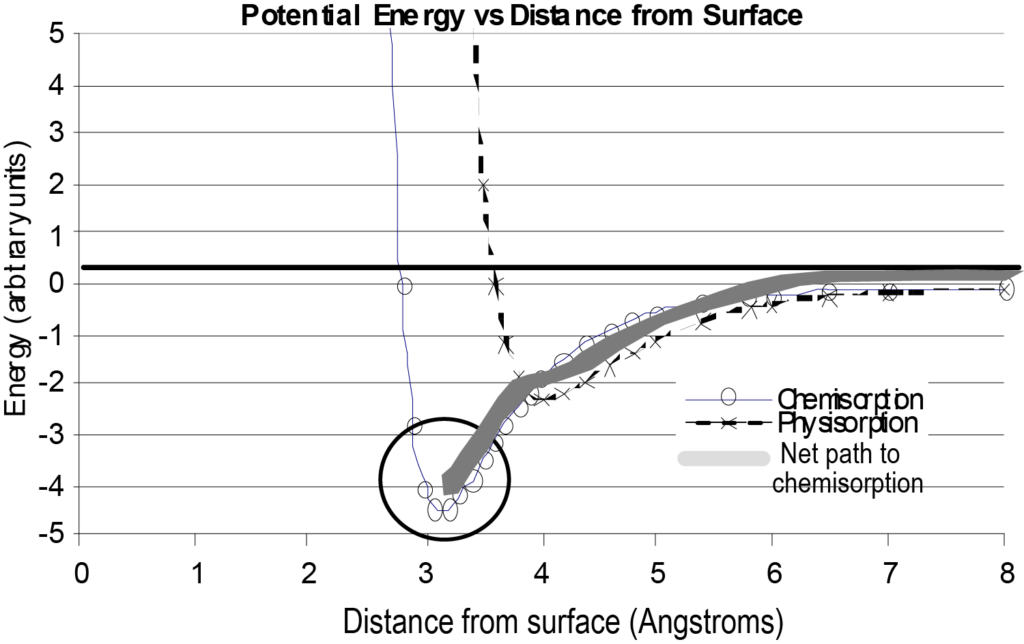
Una situación que cambiaría mucho la forma de la curva de quimisorción de la figura 7 sería que el proceso de adsorción fuera disociativo. La quimisorción disociativa requiere un gasto de energía (energía de disociación) para fracturar la molécula adsorbente. Esto daría lugar a que la trayectoria de la energía neta tuviera un pico positivo justo antes del pozo de energía potencial negativo. Esto se ilustra en la Figura 8. Como en la Figura 8, la trayectoria de adsorción física es inicialmente más favorable (menor energía) a medida que la molécula se acerca a la superficie, pero para disociarse y quimisorberse, se requiere un paso de energía positiva para hacer la transición a la trayectoria de quimisorción. Esta energía es superior a la energía potencial disipada de la molécula.
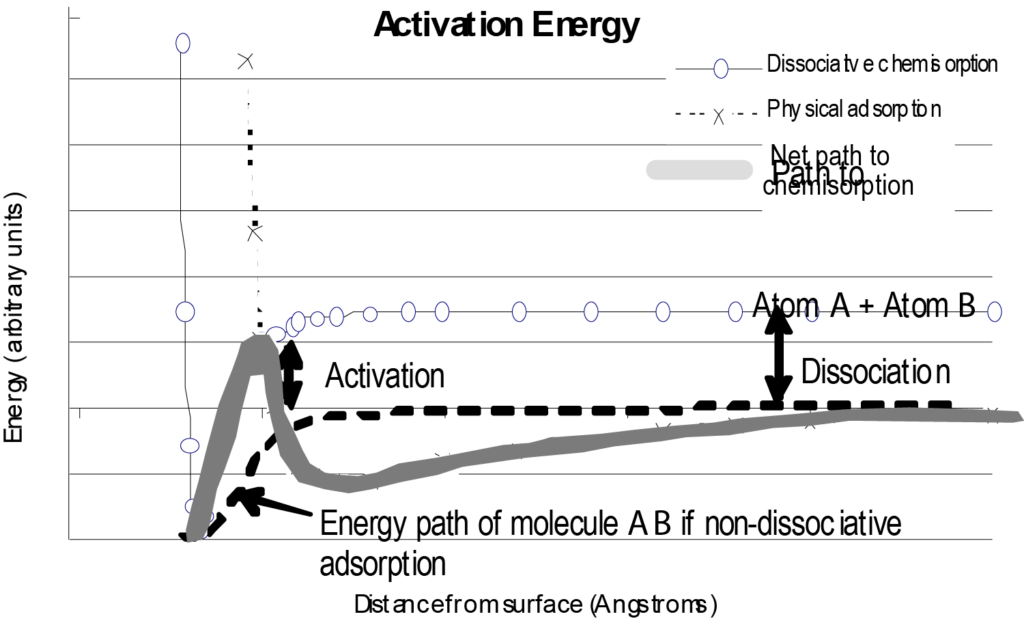
Una variable importante que modifica la forma de la curva de energía de quimisorción no disociativa es el número de moléculas ya adsorbidas en la superficie. Los sitios más energéticos (los pozos de potencial más profundos) se ocupan primero. Esta forma de cobertura continúa hasta que, finalmente, se ocupan los sitios que tienen la energía más baja. La energía trazada en función de la cobertura superficial (carga) caracteriza el grado de heterogeneidad de la energía superficial. Este gráfico proporciona información valiosa sobre la actividad del catalizador en condiciones específicas. Para algunas aplicaciones, en lugar de un gráfico de la distribución de la energía superficial, puede bastar con un único valor de la energía asociada a la reacción.
La adsorción es un proceso exotérmico, por lo que, en última instancia, produce energía(energía de adsorción) aunque se requiera un cierto aporte de energía(energía de activación) para iniciar el proceso. La inversión de la adsorción requiere el aporte de energía, siendo la cantidad requerida la profundidad del pozo de potencial.
Las figuras 7 y 8 se refieren a la energía potencial de una molécula aislada, pero es el potencial energético de todas las partículas del sistema el que determina si el sistema está en equilibrio. En equilibrio, una molécula en cualquier punto del sistema debería experimentar el mismo potencial energético; esto se aplica a las moléculas en el gas o vapor que rodea al sólido y a las moléculas adsorbidas en la superficie del sólido.
Para un sistema adsorbato-adsorbente específico, el equilibrio de adsorción se alcanza mediante un delicado equilibrio entre la presión P de la fase gaseosa a granel, la temperatura analítica T y el grado de cobertura de la superficie ϑ. Experimentalmente, uno de estos parámetros se mantiene constante, al otro se le asigna un valor y el tercero se observa para determinar su valor en el equilibrio. Una isoterma expresa el grado de cobertura superficial en función de la presión a temperatura constante (ϑ = f(P)T), una isobara de adsorción expresa el grado de cobertura superficial en función de la temperatura a presión constante ϑ = f(T)P), y una isostérica muestra la relación entre la presión y la temperatura para un grado de cobertura constante. (P = f(T)ϑ).
La ecuación de Clausius-Clapeyron expresa el calor de adsorción a un grado específico de cobertura superficial ϑ en términos de presión y temperatura, por lo que da como resultado el calor isostérico de adsorción qst. La ecuación en forma diferencial parcial es
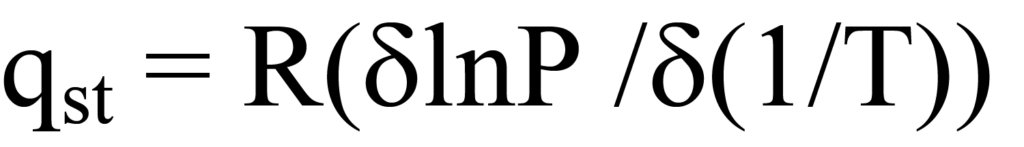
donde T es la temperatura, P la presión y R la constante de los gases.
La obtención de valores relacionados de temperatura y presión para un determinado grado de cobertura se consigue obteniendo múltiples isotermas de la misma muestra a diferentes temperaturas y escalando el eje de la cantidad adsorbida en unidades de grados de cobertura. Esto permite extraer del conjunto de isotermas un conjunto de isobaras trazadas como grado de cobertura frente a la temperatura en el intervalo de temperatura utilizado en los análisis. Del conjunto de isobaras, se puede extraer un conjunto de valores de presión frente a temperatura para varios valores de volumen adsorbido. Éstos se trazan como ln(P) frente a 1/T.
La ecuación de Clausius-Clapeyron, anterior, puede reagruparse en forma lineal, y = m(x), dando como resultado
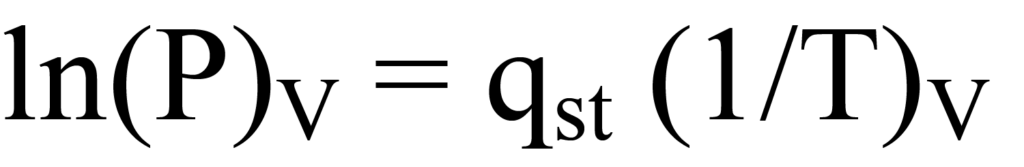
R es una constante de escala que no afecta a la pendiente. Para el valor de la cantidad adsorbida, hay una pendiente diferente, qst. Al trazar el conjunto de pendientes en función de la cantidad adsorbida se obtiene el buscado gráfico de energía superficial frente a cantidad adsorbida, que se relaciona con la cobertura o carga superficial.
El calor de adsorción también puede calcularse a partir de los datos obtenidos por quimisorción dinámica mediante lo que a veces se denomina método de "variación de la velocidad de calentamiento". Puesto que la cantidad adsorbida en función de la presión no se controla por el método dinámico, debe buscarse una expresión de la energía distinta de las de las ecuaciones 10 y 11. Como ejercicio, tal expresión se deduce en los párrafos siguientes. Como ejercicio, en los párrafos siguientes se deduce tal expresión. El resultado (ecuación 21) puede tomarse y utilizarse como un ejercicio de fe, evitando así la pesadez de vadear las matemáticas. Sin embargo, la derivación paso a paso se presenta aquí porque puede ser de interés para algunos lectores y es difícil encontrar una derivación detallada en la literatura. Parte de lo que sigue se ha extraído de los trabajos de Schroeder y Gottfried, Houston, Nix y Garrett.
La desorción implica la tasa de cambio temporal del número de moléculas adsorbidas N, expresada algebraicamente como ΔN/Δt. Esto también puede verse como la tasa de cambio temporal en el número de sitios de adsorción disponibles Na.
(Nota: Los cambios dependientes del tiempo y de la temperatura se tratan más cómodamente desde el punto de vista matemático considerando que cada paso de cambio en el tiempo o la temperatura es infinitesimalmente pequeño. De este modo, DN/Dt puede sustituirse por ΔN/Δt y la manipulación algebraica se sustituye por cálculo diferencial. Para los lectores no familiarizados con el cálculo, cualquier expresión de la forma dX/dY puede considerarse la pendiente de un gráfico de X frente a Y en un valor dado de X).
La expresión cinética para la velocidad de desorción Rd es
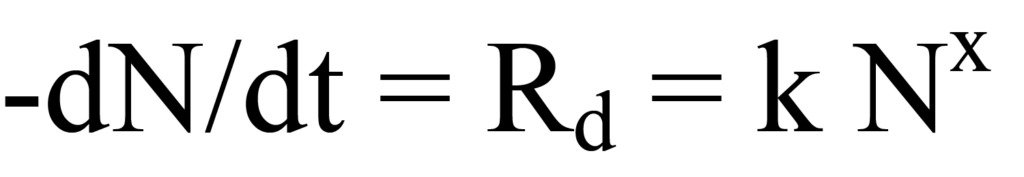
donde k es la constante de velocidad, -N la concentración superficial instantánea (número) de las especies adsorbidas (negativo indica disminución), y x el orden cinético. Aquí sólo se considera el caso más sencillo, la cinética de primer orden (no disociativa); por lo tanto x=1. Obsérvese que la velocidad de desorción cambia constantemente a medida que disminuye el número de moléculas que permanecen en la superficie.
Las figuras 8 y 9 ilustran que la energía de activación puede o no ser necesaria para que se produzca la adsorción. Sin embargo, estas figuras también ilustran que siempre existe una barrera de activación que debe superarse para que la molécula se desorba, es decir, para que la reacción quimisorción-superficie se invierta. La relación entre la constante de velocidad y la energía en reacciones que dependen de la energía, como la desorción, puede expresarse en forma de Arrhenius, en concreto
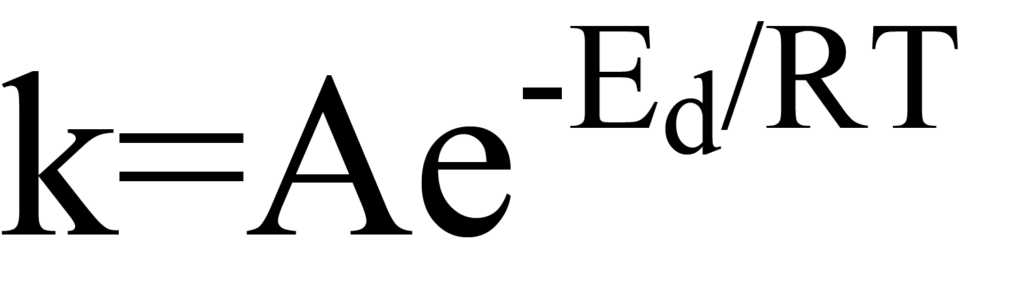
donde Edes es la energía de activación y el subíndice "d" identifica el caso particular como el de desorción. El factor preexponencial A puede considerarse el número de intentos de la molécula por unidad de tiempo para escapar del pozo de potencial. T es la temperatura (grados Kelvin) y R la constante universal de los gases. El producto RT es la energía térmica, por lo que el exponente E/RT es la relación entre la energía de activación y la energía térmica. Mientras E sea mayor que RT, hay poca probabilidad de desorción.
Sustituyendo la expresión de Arrhenius en la Ec. 8 se obtiene
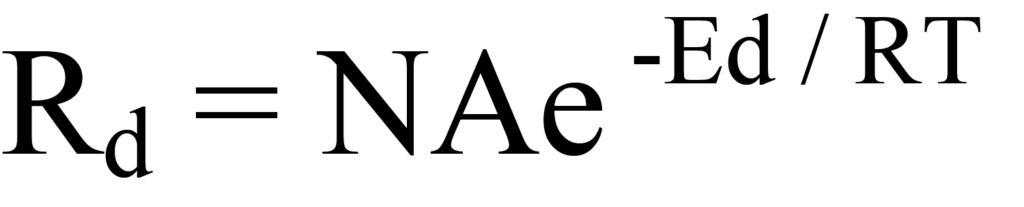
Ésta es la ecuación de Polanyi-Wigner para la velocidad de desorción de primer orden. En la figura 10 se representa cómo cambian la velocidad de desorción (Rd), la cobertura (N) y la relación de energía (Ed/RT) en función de la temperatura, normalizando los valores en el eje vertical para mayor comodidad. Durante un experimento real, la salida del detector correspondería a la velocidad de desorción. La temperatura Tm a la que la tasa de desorción es máxima se observa fácilmente en la Figura 10, como se vería en un gráfico de la señal del detector (cantidad de especies moleculares evolucionadas) en función de la temperatura.
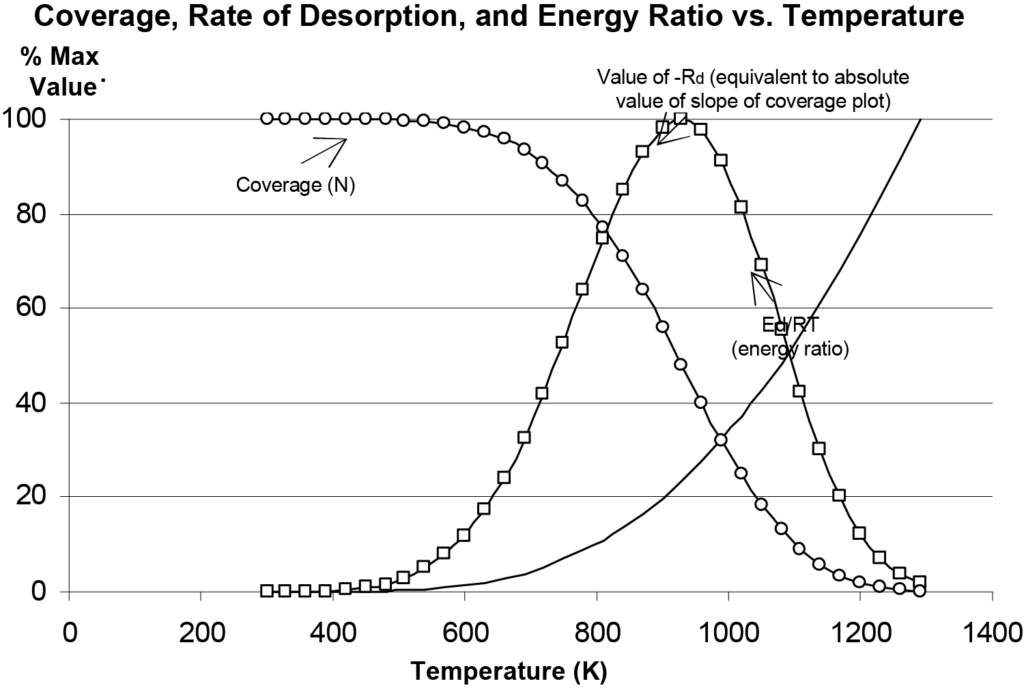
Nota: La Figura 10 fue producida por Microsoft Excel® utilizando la Ecuación 14 y valores arbitrarios para los parámetros. Esto permite experimentar con los parámetros y comparar las distintas gráficas predichas por el modelo de Polanyi-Wigner. Se puede observar que los picos son algo asimétricos alrededor de Tm, aumentando el valor de cobertura inicial hace que el pico aumente en amplitud pero con Tm permaneciendo constante. Aumentar el valor de A hace que el pico se desplace hacia temperaturas más bajas. Aumentar Ed hace que el pico se ensanche y que el valor máximo se produzca a temperaturas más altas.
Extrayendo el logaritmo de ambos lados de la ecuación 10 se obtiene
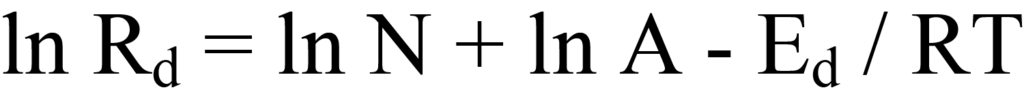
Agrupando esta ecuación en forma lineal, y = (m)x +(b) se obtiene
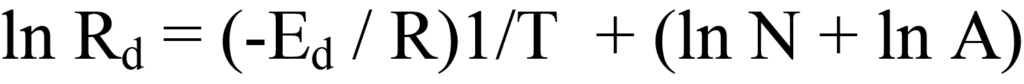
Al trazar ln Rd frente a 1/T se obtiene una línea recta con pendiente -Ed/R e intercepción (ln N + ln A). En este caso, el problema es que no se dispone de valores numéricos para Rd, por lo que hay que ampliar la derivación.
Durante un ensayo de análisis programado por temperatura, la muestra se calienta linealmente, de modo que la temperatura T en cualquier momento t puede calcularse mediante
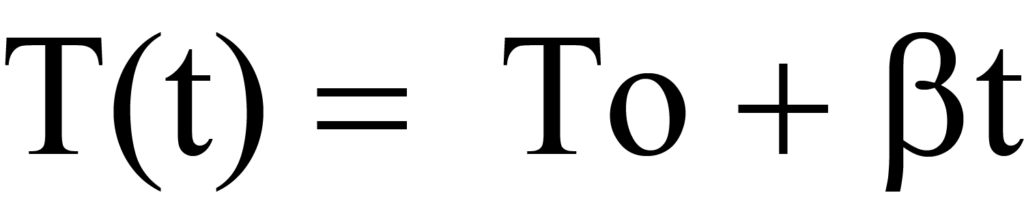
o
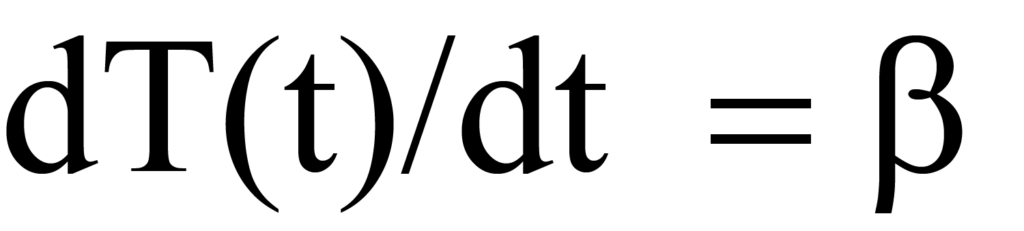
donde β es la velocidad de calentamiento o velocidad de rampa dT/dt en unidades de grados K por unidad de tiempo. A medida que aumenta la temperatura, se produce un cambio en el número de moléculas que se desorben por unidad de tiempo, expresado por dN/dT. Multiplicando la velocidad de calentamiento dependiente del tiempo (dT/dt) en grados/min por la velocidad de desorción dependiente de la temperatura (-dN/dT) en moléculas/grado) se obtiene la velocidad de desorción dependiente del tiempo, Rd. Matemáticamente,
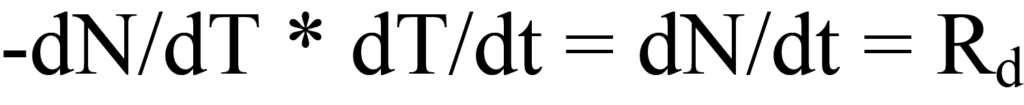
La velocidad de calentamiento β se definió en la Ecuación 15a como dT/dt, por lo tanto, la sustitución en el lado izquierdo de la Ecuación 12 da como resultado
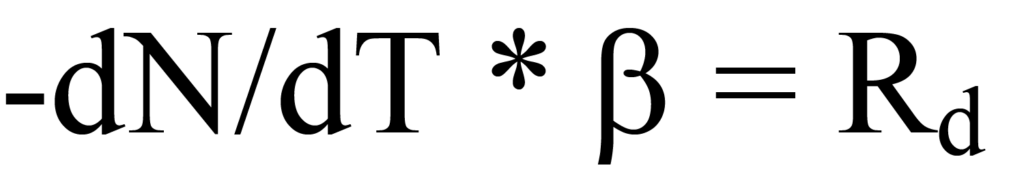
La sustitución de la Ec. 17 en la Ec. 14 da como resultado
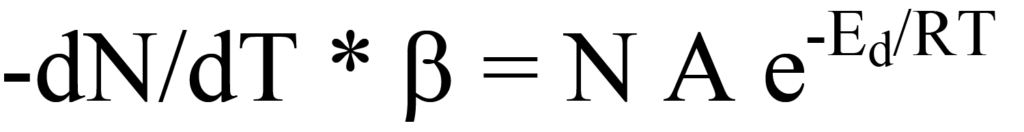
y el reordenamiento produce la tasa de desorción dependiente de la temperatura,
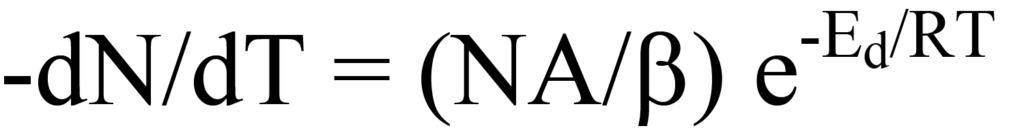
que describe el gráfico de la velocidad de desorción de la figura 10. A medida que aumenta la temperatura, la tasa de desorción se maximiza a Tm y a esa temperatura la pendiente (primera derivada) de la Ec. 15 es igual a cero, es decir,
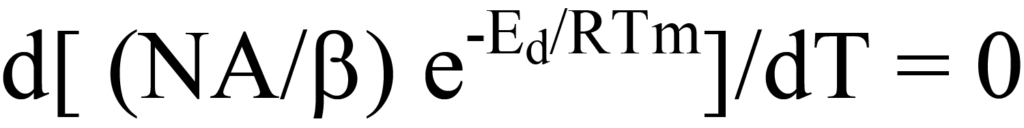
Para mayor claridad y continuidad, se recuerda al lector que la ecuación 20 tiene la forma d(uv)/dT, donde u = (A/b)N, v = e-Ed/RT, y N es una función de la temperatura (T).
La ecuación 20 da como resultado
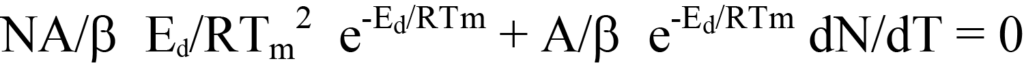
Ahora, sustituye el lado derecho de la Ecuación 19 por dN/dT (un valor negativo; ver Ecuación 12) en la Ecuación 13 y factoriza. El resultado es
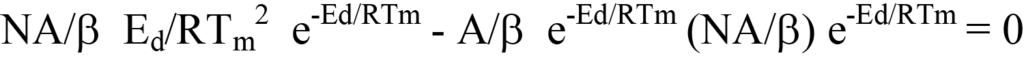
o
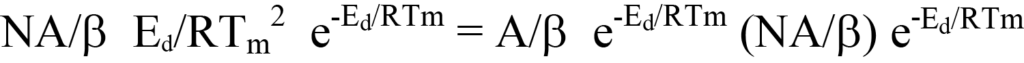
Si se eliminan los términos comunes de los lados izquierdo y derecho, queda

Los valores de Tm, β y R son conocidos; A es desconocido al igual queEd, el valor que se busca. Un método para determinar las incógnitas consiste en expresar los términos de la ecuación 23 en forma lineal, y = mx + b. Esto puede hacerse reordenando en
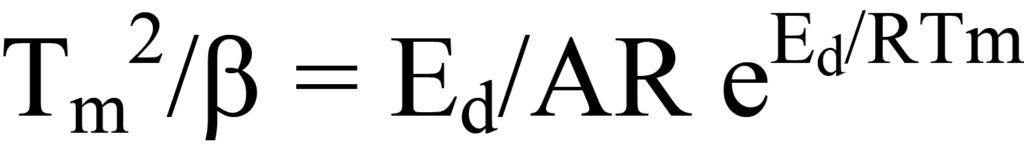
Tomando el logaritmo de cada lado se obtiene

una ecuación lineal en la que y = ln Tm 2/β, x = 1/Tm, m =Ed/R, y b = lnEd/AR. El trazado de y frente a x para una serie de valores de β permite determinarEd. Otra forma de la ecuación 25a que a veces se encuentra en la bibliografía resulta de expandir los términos logarítmicos para obtener
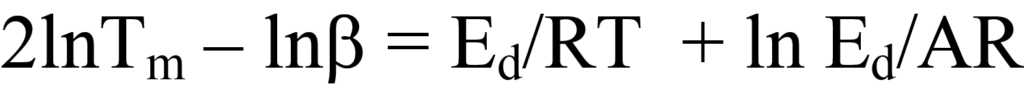
Otra forma de llegar a la Ecuación 25 a partir de la Ecuación 22a es factorizar y reordenar esta última ecuación, obteniendo
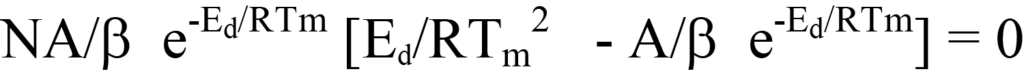
Dividiendo ambos lados de esta ecuación por la expresión entre paréntesis se obtiene
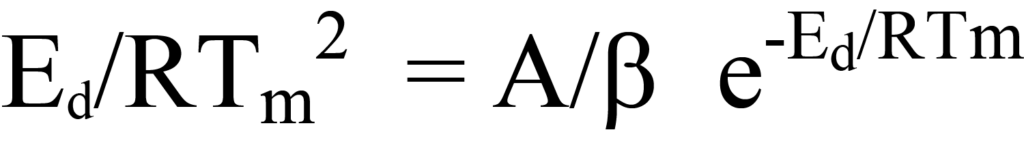
que es igual a la ecuación 23.
También está el caso
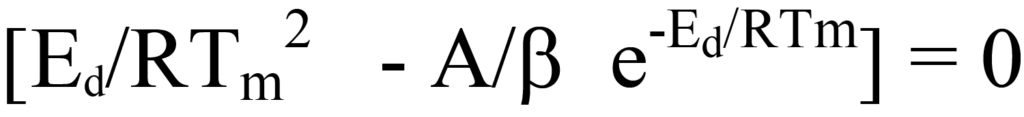
o
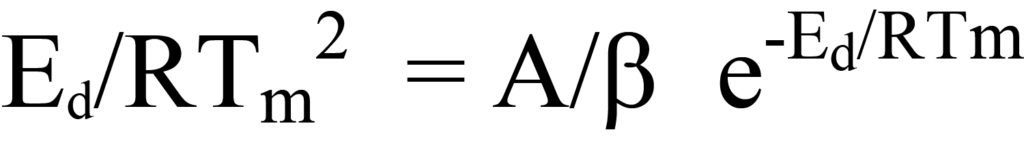
establecido dividiendo ambos lados de la Ecuación 26 por los factores fuera de los paréntesis o deduciendo lógicamente que, puesto que ni el factor NA/β ni e-Ed/RT tienen valor cero durante el proceso de desorción, la expresión entre paréntesis de la Ecuación 26 debe ser igual a cero. Reordenando se obtiene
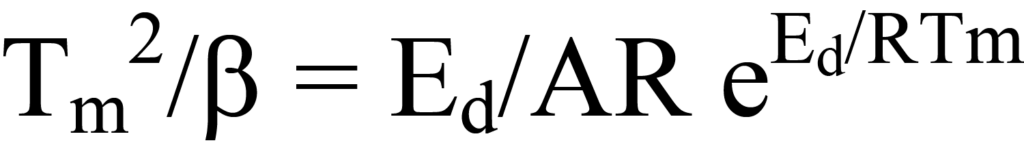
que es igual a la ecuación 24.
Siguiendo el método de Redhead, la ecuación 25 se reordena como sigue:
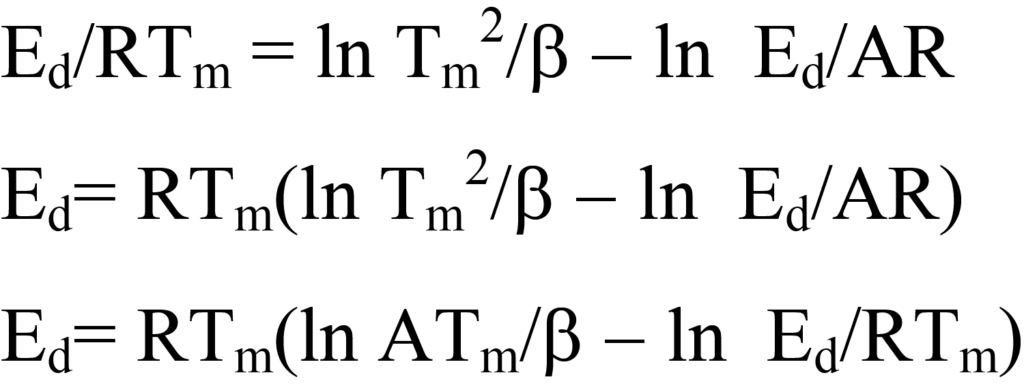
Esta ecuación se simplifica estimando el término ln Ed/RTm del lado derecho en 3,64, lo que introduce un error de menos del 1,5% para valores de A/β entre108/K y1013/K, siendo A típicamente aproximado para cinética de primer orden en1013/s. Así pues,
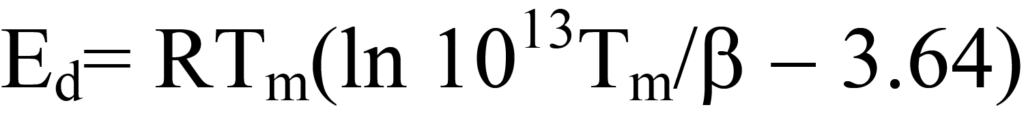
Al igual que el método de punto único para determinar la superficie BET, este método introduce cierto error, pero permite estimarEd a partir de un único cromatograma de desorción. (Schroeder y Gottfried). En lugar de estimar el valor de A, se podría obtener una serie de cromatogramas para diferentes valores de β y determinar los valores deEd y A a partir del trazado lineal.
Al utilizar el método de Redhead, debe recordarse que sólo se aplica a la cinética de primer orden. Con respecto a las reacciones de orden superior, Stoltz advierte (Stoltze): "Describir E obtenida de esta manera como una aproximación es un eufemismo, completamente errónea es una mejor descripción."
Resumen
Los catalizadores sólidos desempeñan un "papel entre bastidores" a la hora de hacer asequibles e incluso posibles muchos de los beneficios que se disfrutan en la vida cotidiana. La quimisorción es un paso fundamental en las reacciones catalíticas, por lo que poder estudiar el proceso de quimisorción a pequeña escala en el laboratorio resulta muy beneficioso. Los modernos instrumentos analíticos capaces de caracterizar estas reacciones son poderosas herramientas para supervisar y controlar la fabricación y aplicación de catalizadores y predecir las características de rendimiento de los materiales catalizadores de nuevo desarrollo.
Recursos
James P. Olivier, Thermodynamic Properties of Confined Fluids I: Experimental Measurements of Krypton Adsorbed by Mesoporous Silica From 80K to 130K, The 2nd Pacific Basin Conference on Adsorption Science and Technology, University of Queensland, Brisbane, Australia, mayo de 2000.
Sven L.M. Schroeder y Michael Gottfried, Temperature- Programmed Desorption (TPD) and Thermal Desorption Spectroscopy (TDS), Universidad Freie, Berlín (Internet http://userpage.chemie.fuberlin. de/~pcfp/V18/pdf/v18.pdf).
Paul L. Houston, Universidad de Cornell Cinemática química y dinámica de las reacciones, Ithaca.
Roger Nix, An Introduction to Surface Chemistry, Departamento de Química, Universidad Queen Mary de Londres (World Wide Web http:// www.chem.qmw.ac.uk/surfaces/scc/scat2_4.htm).
Simon J. Garrett, An Introduction to Surface Chemistry (notas de clase) , Universidad Estatal de Michigan.
W.J. Moore, Physical Chemistry, Prentice-Hall, Inc. (1972).
J.H. de Boer, The Dynamical Character of Adsorption, Oxford en Clarendon Press (1953).
G.C. Bond, Heterogeneous Catalysis- Principles and Applications, Claredon Press, Oxford (1987).
John H. Sinfelt, Structure of Metal Catalysts, Review of Modern Physics, Vol. 51, No. 3 (1979).
Thomas H. Maugh, Industry Steps Up Quest for Catalysts, High Technology, agosto de 1984.
Scholten, Pijpers y Hustings, Surface Characterization of Supported and Nonsupported Hydrogenation Catalysts, Catal. Rev.-Sci. Eng., Vol. 27, No. 1, (1985).
A. Baiker, Métodos experimentales para la caracterización de catalizadores. II. X-Ray Diffraction, Temperature- Programmed Desorption and Reduction, Thermogravimetry and Differential Thermoanalysis, International Chemical Engineering, Vol. 25, No. 1 (1985).
Philip Moriarty, Atoms and Molecules at Surfaces, School of Physics & Astronomy, University of Nottingham ( World Wide Web http:// www.nottingham.ac.uk/~ppzpjm/sect4_2.htm).
M. B. Raschke, U. Ho, Chemisorption energy of hydrogen on silicon surfaces, Physical Review B, Volumen 63.
P. A. Redhead, The Ultimate Vacuum, Vacuum 12, 203-211, 1962.
Per Stoltze, Introduction to heterogeneous catalysis- Concepts and calculations, Departamento de Física, Universidad Técnica de Dinamarca.
Gerard P. van der Laan, Kinetics, Selectivity and Scale Up of the Fischer-Tropsch Synthesis, Tesis doctoral, Universidad de Groningen, Países Bajos, 1999.



